緣起
在就讀台大基因體與系統生物學時,開始碰觸生物資訊,因而開始學習R語言,進而慢慢認識這個有趣且開源的程式語言,在2016年初時,參與了DSP智庫驅動所舉辦的資料科學營隊,主旨是學習開源R語言來做數據分析,小組成員背景迥異,有統計學系、計算機科系、計量金融、醫療背景,而我們小組在幾天營期中,討論出了一個有趣的題目:分析並且了解台中友善老人的程度,在搜集了許多資料後,我們使用WHO裡面的Age-friendly guideline來做基底,結合台灣開放政府的資料來建立指標,最後以地理視覺化的方式來呈現,在營隊結束後,為了進一步好好讓我們的成果能更完善,我們決定要投稿一個會議來激勵我們持續的改進,最後選擇了useR!2016的會議來當作我們的目標,一方面,useR會議是R語言會議裡面最重要的,且今年舉辦地在史丹佛大學,非常值得一試,而在準備投稿的期間,我們把分析的對象擴大到全台灣的縣市,也將視覺化的部分調整更美觀。
為何選擇useR!2016會議
在準備要投稿前對於整個R語言的生態圈不是非常理解,在準備的過程才慢慢理解R語言社群中最重要的會議便是每年一次的useR!會議,裡面會有軟件開發者的發表和交流,且會接受各種R語言相關的海報投稿,而相信這會議蠻值得一去的,畢竟學習開源語言最快速的方法便是積極與其他開發者學習交流!
useR!2016的特色
此useR!會議每年的舉辦地和舉辦人都不同,今年舉辦的地方是在著名的史丹佛大學,位於美國的西岸矽谷的心臟地帶,且獲得灣區地方的大公司贊助,如google、microsoft、H2O、teradata、Oracle、datacamp、RStudio、ebay、Alteryx、AT&T,而主軸演講邀請到很多的大師,如S語言的開發者Richard Becker、Donald Knuth、Deborah Nolan、Hadley Wickham等等。
投稿策略的思考
決定要報名useR!2016會議後,便開始研讀useR!2016會議裡面撰寫的細節,其中適合學生等級可以投稿的為lighting talk和poster session,對於投稿的內容和主題需要如何細化內容和方向,就藉由閱讀2015會議的abstract來一窺究竟,而發現大多數投稿的項目可以大致分為:統計方法的精進、開發新的套件、R語言在各領域的應用,我們的項目變很適合在第三個類別來投稿,再來分析投稿上人的資歷和組織,發現我們這組非“博士”以上層級,大概比較適合投稿poster,所以最後變決定以poster的形式來投稿。其中撰寫項目介紹的時候,我們盡量用關鍵字搜尋2015年的資訊,學習裡面的用字遣詞和文章結構,盡量言簡意賅且用字要精準地表達某個領域的關鍵。
投稿時程和內容需求
useR!2016的報名系統主要是用typeform做成的,流程很順暢清楚,主要需要提供:摘要、專題相關的網站、專案的github,這三項是最需要費盡準備的,其中與投稿專題相關的網站,我們使用在營隊裡團隊討論的hackpad,裡面有詳細記錄我們整個從題目發想、工作分工、資料收集、分析結果,專案的github建置則參考網路上各種“分析專案”的資料夾架構,便是用程式碼、分析結果、分析資料,三個大分類來置放整個分析過程產生的資料,準備工作大概花費一個多月,將整個專題內容比較有條理的整理成摘要,變且建置乾淨整潔的github repository。
會議參與的補助:開放文化基金會
當知道投稿通過後,我們開始思考如何獲得補助來參與會議,其實對於這類開源會議的補助,從校方或是教育部方面來著手都會比較困難,一方面是這類補助的規定會比較死,通常都需要找尋指導老師,而我們組成來自各個學校,很難用這種方式來申請,最後找到開放文化基金會裡補助青年參與國際開源會議的計畫,恰巧符合我的需求,開放文化基金會這計畫需要繳交的資料便是:1.參與會議的介紹,和值得參與的理由、2.投稿的內容、2.經費的估算,整個過程非常順利,對於學生參與國外大型開源會議來說是非常好的資源!
在實務面來說,開放文化基金會的申請是遞交後便開始審核,整體對於規劃機票和住宿的費用申請很有彈性,相對於學校或教育部需要以“三個月一季”為單位,將會議開始的時間點來推算申請日程,造成會議開始時間點和得知補助通過的時間點很接近,造成購買機票或是規劃住宿費用提升。除此之外,開放文化基金會要求回國後要舉辦分享會,將在國外的所見所聞分享,而籌備的過程需要自主積極的規劃,而基金會也會提供非常多的意見和幫忙!
useR!2016會議架構
今年的會議議程總共有四天,從6/27到6/30,會議場地遍佈史丹佛校園的各個場館,從台灣轉機直接搭到聖荷西機場,再轉搭接駁公車和火車到史丹佛大學,史丹佛大學的校區就在帕羅奧圖站,從火車站下車其實就到校區,校區裡面有醫院、購物中心,內有大學公車在大學內,住宿方面則因為在灣區的關係非常昂貴,普通旅館幾乎都要一晚上一萬元左右,但會議有提供史丹佛大學宿舍可以申請,費用可稍微節省許多,但名額不多,會先讓講者或是報告者申請,議會內容形式主要有:工作坊、大師講題、小型演講、海報分享等內容,所有的會議內容都有錄影,這次微軟負責直播和錄影,整體的錄影效果非常好,會後已經將影片放置在microsoft的channel 9 平台上,是非常好的學習資源。
會議四天都有提供午餐和點心,第一天以工作坊為主,主題從機器學習、模型建置、統計分析、R工作流管理、RStudio新功能介紹,休息時間則有咖啡甜點供應,第二天到第四天則以大師講座、小型演講、海報演講為主,會議採用Sched平台,四天下來大大小小的演講超過400多場平行議程,如何選擇常常事件令人頭痛的問題,而Sched剛好提供一良好的個人會議議程規劃,可以安排自己有興趣的講題,且可以用地圖的遠近來挑選,才不會發現最後安排的行程不順路,最後會將每個人的會議議程選擇自動匯整成為個人行程,如同google日曆一般,前一天晚上會提醒你隔天要去哪邊,且每一場講座都有清楚的摘要和介紹,另外使用這套系統的有趣之處在於你可以看到其他人的行程安排,同一場會議有誰要去,事前知道哪一場會議是最熱門的,連海報session都可以是先看到誰對這主題有興趣,這種把網路社交的概念帶入會議參與中,可以發揮networking最大化的效果。
useR!會議工作坊
第一天為工作坊時段,總共有18場可供選擇,每場三小時,中間會有一個休息時間,講師皆為資深的開發者,資料科學家、技術叢書作者、教授、線上課程講師,在工作坊前就能看到每個工作坊的事前閱讀和安裝準備文件,大多數講者都有準備好的課程github頁面,將所有需要的東西放在裡面,工作坊的過程底下的參與者也能一同修改內容,互動良好,在教學上,有的講者在AWS上架設RStudio的server,裡面先設置好需要的環境提供給參與者,參與者直接連線到遠端帳號上操作,如此便可以減少安裝環境和依賴包所需的時間(通常現場網速都很慢),讓參與者儘速可以動手,或是使用Jupyter notebook裝上R kernel,在上面展演分析的過程,可以學習他們這種分享的方式,不限於使用簡報而已,更多的與參與者一同演練。
useR!會議的大師講座
這次會議的大師講座剛好串起最近R社群很火熱的主題:如何創造好的開源社群(Richard Becker從S語言的興衰來思考R語言的興盛)、Literate programming(Donald Knuth從其經典著作The Art of Computer Programming談好的程序撰寫)、資料科學的教學風氣(Deborah Nolan以其在伯克萊教學經驗談“好的”資料科學該怎麼教或是學)、Hadley Wickham(針對gg系列未來發展方向的解說),Daniela M. Witten是知名統計學家Rob Tibshirani的“嫡傳弟子”,研究專注於高維度資料的機器學習,尤其是基因組測序資料。
Richard A.Becher是S語言的創造者,在R語言出現之前,S語言發展了將近四十年,Richard Becher是當時貝爾實驗室的研究領導學者,在1970年代貝爾實驗室便是當時統計編程的重要發源地, S語言便是應貝爾實驗室裡頭科學家、工程師們的需求而存在的,此次演講變分享他是如何讓S語言茁壯和當時開發的種種心得,其提到其開發S語言秉持者一些基本原則,第一,其開發的功能都是在貝爾實驗室裡頭所需要的,第二,S語言只有其和少數幾位專家們決議重要的發展發現,第三,其開發跟者當時的unix系統,所以移植性佳,最後Becher也省思了為何S語言會消逝,或許這場演講是為了讓R開源社群的夥伴更理解一個語言的興起和衰弱,是存在哪些因素,並且從其中借鑑。
Donald Knuth是史丹佛的計算機科學教授,開發了目前為人熟知的Tex,其一發布其原代碼幾乎沒有錯誤,他撰寫的編程的藝術(the Art of Computer Programming)在計算機科學裡佔有非常重要的角色,這次他演講的題目literature programming正式R開源社群近年來很重視的議題,如何撰寫更reproducible以及sharable的代碼和工具,所以他就分享他對於literature programming的看法。
Deborah Nolan是柏克萊大學的統計教授,他分享了其對於所謂“資料科學”的教學心得和應該要有的方向,批判了傳統統計學總是以學理公式的方式來傳授,Nolan教授更強調要在“脈絡”下去學習分析資料,從中學習統計的觀念,且搭配者編程語言,讓學生能更“動手”地來學習,最好從資料最“原始”未受清理的階段開始,可以從他的書Data Science in R: A Case Studies Approach to Computational Reasoning and Problem Solving來學習他整套的資料科學教學方式,書中便是一章章使用案例的方式,帶出一個科學思維的脈絡,並從資料最“原始”的狀態開始處理,他的演講可以對於我們學習資料科學有更好的思維準備。
Hadley Wickham是目前R語言界最呼風喚雨的才子,本身是Rstudio的首席科學家,有一群很好的夥伴在開發各式各樣的R語言封包,其中最讓大家知道的是ggplot2,但除此之外,他其實有一整套開發思維,在於如何讓人們能更有效率地使用R在進行資料分析,這次主要帶來其tibble、tidyr、dpylr的想法,整個演講內容提綱挈領,且搭配者操作與台下的互動,相信未來他會開發出更多好用的封包,甚至整個改變了R語言的撰寫風格。
Daniela M. Witten是統計學術界的新星,目前是華盛頓大學統計系的教授,本身專注於處理高維度數據機器學習的方法,跟有名的史丹佛統計學家Rob Tibshiran一起寫過An Introduction to Statistic Learning,是其在史丹佛大學的嫡傳弟子,開發出許多機器學習相關的R語言封包,這次分享其使用Convex Penalties來做回歸分析。
這幾位大師級的分享,串鏈了這幾年社群裡面很火熱的議題,很值得把原始的演講影片調出來欣賞。
(Microsoft channel9平台裡面有這次演講的直播影片)
在眾多的Lighting talk裡面(因為有400多場),有的只有簡短五分鐘,大多數都會分門別類的,比如資料視覺化、生物資訊、醫療數據、資料庫、雲端計算、統計模型、應用等等,大多是開發者分享和介紹自己的項目,希望大家多多使用,這些分享可以清楚讓我們這些R語言終端使用者深深體會到其實我們並沒有跟開發者距離那麼遠,他們其實很希望使用者們跟他們建立關係,回應需求,甚至提供改善的意見,且也會發現到R語言的封包如此多種,要小心謹慎的選擇可靠的開發者,才不會浪費學習成本,但到頭來開發者停止繼續改善。而優秀的開發者會把使用者遇到的困難當作是自己的問題,努力去降低使用的障礙,比如Hadley便是這種極端偏執於開發出好用的封包。
在第三天下午則是我個人的海報發表時間,場地是在戶外草地搭建的區域,每個人可以提早大約十天寄電子檔給大會幫忙輸出,展位大小約莫全開紙張大,旁邊就是贊助商的攤位,發放各式各樣的宣傳禮品,間接帶動來這邊看海報發表的人群,為了準備好如何介紹發表的內容練習了很多版本,因為每個人有興趣的程度都不一樣,這種練習介紹自己在做什麼的方式蠻棒的,可以很快速地拉近參與者,相對於午餐搭訕,更有正當的話題,同時,也可以觀察每個人海報製作的邏輯都不太一樣,生物背景分享者的海報、計算機科學背景分享者的海報、數學物理背景分享者的海報都不同,大抵是機器學習相關的主題最讓大家有興趣。
這趟的useR!2016會議經驗其實非常的難得,一方面舉辦地點在史丹佛大學,可以趁機會到附近逛逛,體會一下矽谷的風情,另一方面,則是會議參與者都是R語言開發者,可以近距離觀察高手們在想什麼,整個R語言社群有什麼很熱門的話題,檯面上的大人物有誰,在會議過程中可以觀察和學習不同國家的交流方式和開場白,都是很有目的性的溝通,短時間內介紹自己的職位在做些什麼事情,如何談段大家有興趣的話題,避免尷尬,甚至很多參與者在會議前都有書信溝通,要在會議當天見面和討論合作內容,唯有不斷提升自己的專業能力,才有機會在國際舞台上跟更多人溝通和來往,希望這些心得分享能鼓勵更多台灣的夥伴跟開放文化基金會爭取出國補助機會,或是讓對useR!會議有興趣的同好能有更多的參考資訊。
Frances C. Arrillaga Alumini Center為會議的主場館,在這邊註冊,裡面有三個會議場地,分別為MaCaw hal、Barne/McDowell 、Lane, ,此處還有提供午餐和點心的戶外座椅區。

海報在第二十號攤位,第三天的海報發表就站在這海報前分享自己的專題內容,旁邊都是知名資料科學公司的攤位。
在主場館裡的Lane會議廳實景
大師演講系列裡每個教授都有直播服務,避免場館會坐不下所有人。

會議場地McGaw Hall,所有的大師講座會舉辦在這邊。

在Campbell Rehearsal Hall,在另一個區域,R工作坊的舉辦地。

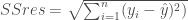 {1}{n-1}\sum_{i=1}^{n}(X_i – \bar x)(y_i – \bar y)$
{1}{n-1}\sum_{i=1}^{n}(X_i – \bar x)(y_i – \bar y)$









