這邊分享一下最近一個清洗資料的流程,做生物資訊的雜活其中一個重要的事情就是把來自各方的資料整合在一起,許多細節是必須把手弄髒才知道的!
問題
最近幫忙解決的這個問題是科內用來出次世代定序報告系統的某個資料表,自從2016年的助理離職,就沒有人能幫忙更新,這也是臨床檢驗科室的問題,老店員工資深,很難有人願意重新投入學習寫程式清資料等等,老闆也不一定知道這箇中的重要,不過相信這是可以慢慢改變的。
這個資料表暫稱Hotsopt.bed,是用來幫忙次世代定序之Panel在定序後的生物資訊流程中作為資料註解的資料表,表格長得如下面這樣:

這是用來幫助次世代分析流程中,當遇到定序品質不好的區域,能把此區域已知具有臨床意義的變異點給吐出來。整個資料表很單純,第一欄是染色體位置、第二和第三欄是這個位置的開始和結束、第四欄是跟這位置相關的臨床位點ID(舊版本是用rs ID,我接手後應該會改成clinvar ID,比較貼近需求)、第五欄則是綜合這個位置的變異資料以及變異起始前一位的序列,最後一欄則是這區域為在哪個target panel的Amplicon。這個表格目前只想放在clinvar DB上面已經住解為Pathogenic和Likely Pathogenic的位點。
該串接的資料庫: NCBI clinvar, dbSNP
從上面的問題,可以知道工作重點是:建立能定期下載特定基因列表的clinvar註釋,然後在看這些位點為在哪個panel的amplicon,以及此位點的序列變化和從參考人類基因組抓出變異位點起始位置前一位的核酸序列,最後整合成hotspot的格式。
因此需要串接的資料庫為
- NCBI: clinvar, dbSNP
第一步:使用NCBI Entrez Direct檢索基因列表最新在clinvar的位點資訊
相信這邊的做法有蠻多的,這邊因為前陣子因為想幫科內建立血庫分子資料庫,所以摸了一下NCBI 的Entrez Direct,就順勢使用這個工具,Entrez Direct 是美國國家生物技術資訊中心(The National Center for Biotechnology Information, NCBI)所提供的一個命令行工具,讓人可以直接使用Unix terminal的方式來針對NCBI內的各個資料庫。基本上就是下面那個下拉表單所提供的所有資料庫都可以調用。(超級佛心的,幾個代碼就可以調用全世界最大的生物資料庫QQ)

這個Entrez Direct(EDirect)可以有幾個安裝方式,可以由Anaconda,也可以直接下載後安裝。 (之前有幾篇文章介紹這個命令行工具 Pubmed E-utilities API 使用規範, 使用python來調用pubmed API快速整理文獻, Pubmed API介紹) ,安裝完Entrez Direct後,他會有幾個命令行的函數是可以用的:
- esearch
- elink
- efilter
- efetch
- xtract
- einfo
- epost
- nquire
藉由組合上面這幾個指令,便能擁有調用NCBI資料的技能,非常實用!且相關的說明文檔有越來越好,相對於幾年前。
這邊所使用的關鍵語法是
for gene in $gene_list
do
echo download $gene
echo ==========================
echo " "
esearch -db clinvar -query $gene[gene]|
efetch -format docsum|
xtract -pattern DocumentSummary -def "-"\
-element Id object_type title\
-block variation_set -subset variation -element measure_id cdna_change\
-block variation_set -subset assembly_set -if assembly_name -equals "GRCh37" -def "-" -element chr start stop \
-block clinical_significance -def "-" -element description last_valuated \
-block genes -subset gene -if symbol -equals "$gene" -def "-" -element symbol GeneID strand >> gene_clinvar.txt
sleep 2s
echo =============== down =======
done
上面的代碼主要是使用三個指令:esearch、efetch和xtract,先用esearch去查詢NCBI內對於單一個基因的最新資料,在使用efetch來下載,最後使用xtract來把資料做前處理來整理成比較。這邊需要花一些的坑就是去調整xtract的pattern,看我們需要什麼資料做後續的處理,然後針對這些資料來做pattern的設計。

每一行都是在這個基因區域中,相關的變異位點資訊,包含其編碼、變異種類、變異細節、其臨床註釋之意義等等。
接者就可以把這個資料去往下做更新。
第二步:將從NCBI下載的資料進行清洗,只留下位點具有Pathogenic或Likely Pathogenic標注的
拿到這些資訊後,接者便是應用各種技巧來進行整合。(資料處理的坑就是這樣,不過也是有趣的地方,每次都會多學到一點點不同的代碼寫法和狀況)
下一步,我們先用R來把這個資料讀進去後,只留下Pathogenic和Likely pathogenic的位點資訊,聽起來簡單,但下面是真實情況的資料:

上面是看各個原始資料中,臨床位點的注釋,會發現很多奇怪的狀況,這邊就是一些清資料的活,確認這些不一致的來源,並且將其處理成乾淨的版本,去掉許多注釋可能沒有完成的位點資訊,最後只留下相關的位點。這邊可以善用dplyr的強大功能搭配字串處理的stringr,我這邊則是使用mutate搭配case_when來做處理。
第三步:利用clinvar ID, allele ID來彙整資訊
取得新更新的特定基因列表中的變異位點相關的ID資訊後,可以以此為參考,來跟不同資料來做收集。尋這些位點分別是在Ion的哪個 Amplicom上、過濾clinvar VCF檔案取得ref和alt資料、使用位置資訊來從reference genome fasta來取得前一位置的位點序列。
這邊使用clinvar ID, allele ID和其相關的位點來取得下面三個資料
- 取得每個變異位點在Amplicon的位置上 | 使用bedtools
- 取得每個變異位點前一個序列的核酸資料|使用seqkit和seqtk
- 取得每個變異位點的實際資訊|使用VCFtools
這邊可以借助Bedtools 來做兩組不同位點資訊的比較,比如位點是否位在特定位置區域上等的分析。

比如這邊我需要做的就是每個位點實際位在的Amplicon位置,這邊就必須要拿位點資料和Amplicon資料來做處理。
bedtools intesect -wb\
-a CRC_clinvarBed -b CRC_amplicon_bed|\
cut -f 1,2,3,4,5,6,10,14
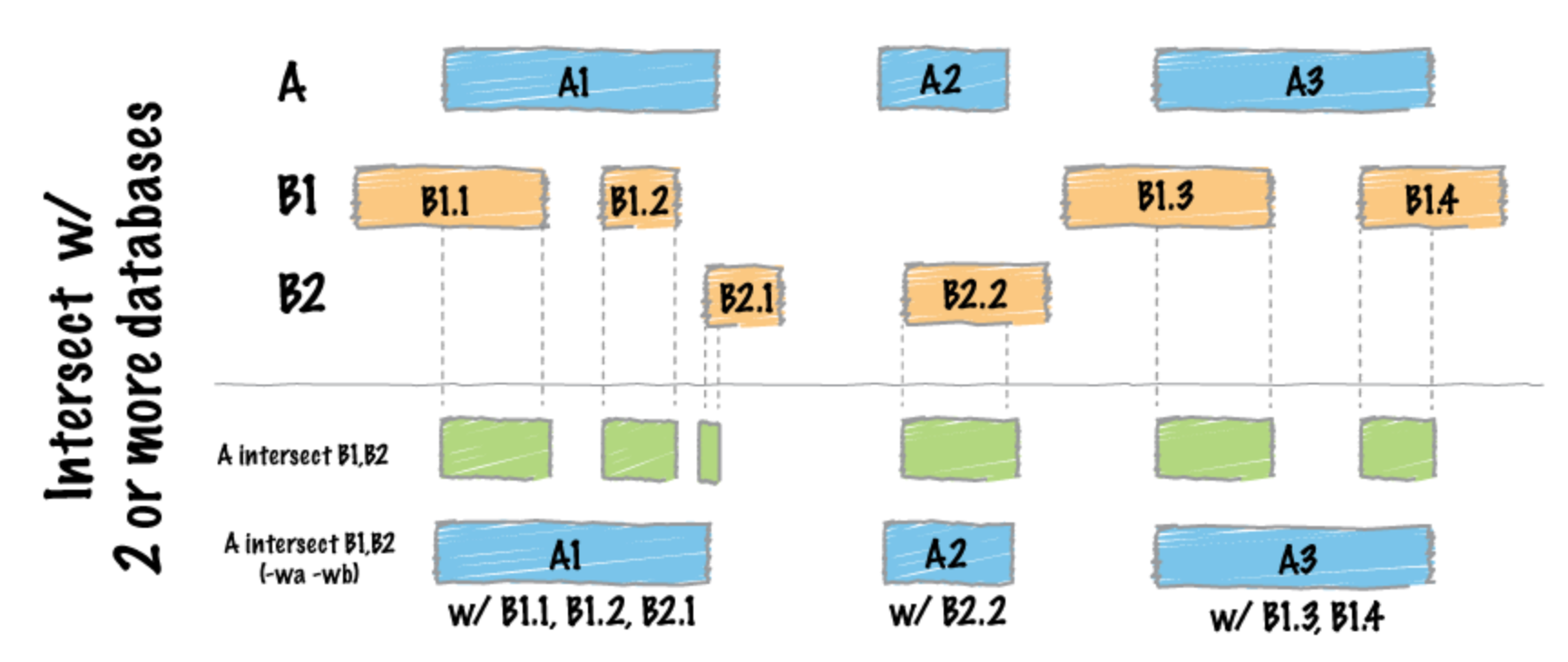
這邊代碼的意思是去看每一個-a後面的檔案其在-b後面的amplicon範圍,並且將兩個資料合再一起,並且最後使用bash的指定cut,只留下特定想要的欄位供後續的使用。像是下方的示意圖:
另一部分,想要取得特定位置的序列資訊,則可以使用seqkit這個工具,他可以吃bed檔案格式來吐出相關的序列資訊,也可以使用seqkit來清理從NCBI所下載。這邊可以直接去NCBI的ftp下載參考序列組,其資料夾長得如下:

下載後,可以看到其實際fasta檔案有非常多細節,有297個片段(第一次抓參考基因組,通常都以為只有24個染色體片段,實際上每個染色體除了主要資料外,還有很多“補丁”的片段)。
seqkit是一個可以用來取代seqtk的分析命令行工具,可以用來處理和分析fasta和fastq的檔案格式,這樣的的代碼和sed來做清洗,最後只留下23對染色體的參考序列,並且使用seqkit來取變異點前一位置的序列。下面的代碼也有用到seqtk,用來提取特定的染色體序列出來。
P.S: 這邊有一個要小心的坑,使用seqkit subseq來擷取序列時,用bed檔或是直接下指令,兩者的索引會是不同的。
seqkit seq -n GRCh37_latest_genomic.fna.gz | grep "GRCh37.p13 Primary Assembly"| grep "NC" > primaryAssemble.lst
# only use primary assembly hg19 reference genome
seqtk subseq GRCh37_latest_genomic.fna.gz primaryAssemble.lst > onlychr_GRCh37_latest_genomic.fna
# rename the hg19 reference header and preprocess of the title
sed '/^>/d' file.fa | wc -l
sed 's/NC_.* Homo sapiens //' draft_o_onlychr_GRCh37_latest_genomic.fna > draft_1_onlychr_GRCh37_latest_genomic.fna
sed 's/, GRCh37.p13 Primary Assembly//' draft_1_onlychr_GRCh37_latest_genomic.fna > draft_2_onlychr_GRCh37_latest_genomic.fna
# get the reference sequence from bed file
seqkit subseq --bed $CRCRefAnchorBed draft_2_onlychr_GRCh37_latest_genomic.fna|\
sed 'N;s/\n/\t/' |\
sed 's/^>//' |\
sed 's/\.//' |\
sed 's/://' > $CRCrefAnchor_file
# get the reference sequence from bed file
seqkit subseq --bed $EpilepsyRefAnchorBed draft_2_onlychr_GRCh37_latest_genomic.fna |\
sed 'N;s/\n/\t/' |\
sed 's/^>//' |\
sed 's/\.//' |\
sed 's/://' > $EpilepsyAnchor_file
接者,為了取得每個VCF檔案裡面的序列變異細節,會使用VCFtools,這邊也是踩了一個軟體的坑,在VCFtools裡面的snps篩選,所使用的其實是clinvar ID而非snpID,這邊真的是頗坑的。
vcftools --gzvcf $vcf_file --snps $Epilepsy_VCF_Bed --recode --recode-INFO-all --out 20210717_epilepsy_annotation_VCF
這個代碼的參數意思:–gzvcf是輸入壓縮過的vcf檔案,–snps則是以提供每行一個clinvarID的列表,用來篩選落在這標單的位點資訊。
第三部:把全部資料彙整成最終檔案
最後的部分可以在R裡面處理,比較high level的資料處理在比較小的資料集中使用R相對方便,因為前面三個檔案都是以tab間隔之檔案,所以讀進去後可以在R裡面彙整成最終的檔案格式。可以用到dplyr裡面的rename、left_join、filter、mutate來做最終的匯聚。
outputCRC_clinvarBED %>%
dplyr::rename('chr'=V1, 'start'=V2, 'end'=V3, 'clinVarID'=V4, 'alleleID'=V5, 'dbSNP'=V6, 'Amplicon'=V7, 'Gene'=V8) %>%
dplyr::left_join(., CRC.annotation.VCF, by='clinVarID') %>%
dplyr::mutate(Anchor_pos=paste0(chr, '_', as.character(start-1))) %>%
dplyr::left_join(., CRC.annotate.ref.anchorfile, by='Anchor_pos') %>%
dplyr::filter(!is.na(REF)) %>% dplyr::filter(!is.na(AHCHOR)) %>%
dplyr::mutate(Info=past0("REF=", REF, ";OBS=",ALT, ";ANCHOR=", ANCHOR))
清資料的過程蠻多小細節的,真的是每次都是全新的坑,只是越清會越知道什麼坑是小坑,什麼坑是大坑,以及比較熟悉各種基因資料的格式比如bed format, VCF format和各個資料庫中的關聯性。
