network analysis就跟任何種類的分析一樣,大部分的時間會花在“clean data”,也就是把資料處理成適合分析的格式。而在network analysis中,關鍵是節點(nodes)和節點間的關係(edges),而非一般我們用observation為row,variable為column的形式。
有兩種常用的方式,來記錄一個網絡,那便是sociomatrix和edge list。
sociomatrix
- 將所有的node之間的關係用一個巨大matrix來記錄,row代表是starting node,column代表是receiving node,但當這個network很巨大的時候,此種形式大部分的cell都會是空值。但資料小時候很一目了然。

edge list
- 直接用一個table記錄每個paired的關係,第一個column代表是starting node,第二個column則是receiving node,這種方式比較直覺,在資料量大的時候處理速度也會比sociomatrix建立的網絡還快,因為少去了很多empty data。

除了單純定義好一個網絡中的node和node之間的關聯,最重要的當然是要把“意義”放進去,就是所謂的屬性(attribute),比如這個網絡是在看臉書好友網絡,而每個node屬性就要被記錄有人、身高、體重等等,而node之間的edge也會有強與弱的分別,是深交或是一般朋友,這些就是要儲存進入網絡資料之中的。
用簡圖來描述R裡面一個R network object要有的資訊
種類 描述
Nodes 網絡中的節點
Ties 節點間的關聯
Node attribute 節點的屬性
Tie attribute 網絡中關聯的屬性
Metadata 整個網絡的相關資訊
在R中創建一個network object
<
div>
#load the library
library(statnet)
#Creat a network object with sociometrix data format
netmat1 <- rbind(c(0,1,1,0,0),
c(0,0,1,1,0),
c(0,1,0,0,0),
c(0,0,0,0,0),
c(0,0,1,0,0))
rownames(netmat1) <- c("A","B","C","D","E")
colnames(netmat1) <- c("A","B","C","D","E")
net1 <- network(netmat1,matrix.type="adjacency")
class(net1)
summary(net1)
##Visualization
gplot(net1)
gplot(net1, vertex.col = 5, displaylabels =TRUE)
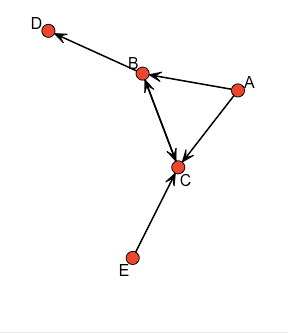
#Creat network object with edge list format
netmat2 <- rbind(c(1,2),
c(1,3),
c(2,3),
c(2,4),
c(3,2),
c(5,3))
net2 <- network(netmat2, matrix.type="edgelist")
network.vertex.names(net2) <- c("A","B","C","D","E")
summary(net2)
#Transform between the differnet data structure
as.sociomatrix(net1)
class(as.sociomatrix(net1))
all(as.matrix(net1) == as.sociomatrix(net1))
as.matrix(net1,matrix.type = "edgelist")
#-Managing Node and Tie attributes
#Node attribute
set.vertex.attribute(net1, "gender", c("F","F","M","F","M"))
net1 %v% "alldeg" <- degree(net1)
list.vertex.attributes(net1)
summary(net1)
get.vertex.attribute(net1,"gender")
net1 %v% "alldeg"
#Tie attribute
list.edge.attributes(net1)
set.edge.attribute(net1,"rndval",
runif(network.size(net1),0,1))
list.edge.attributes(net1)
summary(net1 %e% "rndval")
summary(get.edge.attribute(net1,"rndval"))
#example for the value netwrok and modify the edge attribute
netval1 <- rbind(c(0,2,3,0,0),
c(0,0,3,1,0),
c(0,1,0,0,0),
c(0,0,0,0,0),
c(0,0,2,0,0))
netval1 <- network(netval1,matrix.type="adjacency",ignore.eval=FALSE, names.eval="like") #use the vertex's vaule added to attribute
list.edge.attributes(netval1)
get.edge.attribute(netval1,"like")
as.sociomatrix(netval1)
as.sociomatrix(netval1,"like")
#--------------------------------------------------------------------
#igraph
#detach the statnet during the duplicate of function name
install.packages("igraph")
detach(package:statnet)
library(igraph)
#create from sociomatrix
inet1 <- graph.adjacency(netmat1)
class(inet1)
summary(inet1)
str(inet1)
#create from edge list
inet2 <- graph.edgelist(netmat2)
summary(inet2)
#modify the attribute
V(inet2)$name <- c("A","B","C","D","E")
E(inet2)$val <- c(1:6)
summary(inet2)
str(inet2)
#--------------------------------------------------------------------
#Going back and forth between statnet and igraph
install.packages("intergraph")
library(intergraph)
class(net1)
net1igraph <- asIgraph(net1)
class(net1igraph)
str(net1igraph)
#Imporintg Netwrok data
detach("package:igraph",unload=TRUE)
library(statnet)
netmat3 <- rbind(c("A","B"),
c("A","C"),
c("B","C"),
c("B","D"),
c("C","B"),
c("E","C"))
net.df <- data.frame(netmat3)
net.df
write.csv(net.df, file = "MyData.csv",
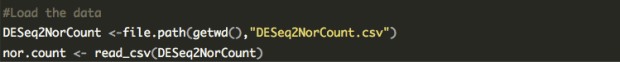
row.names = FALSE)
net.edge <- read.csv(file = "MyData.csv")
net_import <- network(net.edge,matrix.type = "edgelist")
summary(net_import)
gden(net_import)
#Common Network Data Task
#Filtering Networks Based on Vertex or Edge Attribute Values
n1F <- get.inducedSubgraph(net1,
which(net1 %v% "gender" == "F"))
n1F[,]
gplot(n1F,displaylabels = TRUE)
deg <- net1%v% "alldeg"
n2 <- net1 %s% which(deg >1)
gplot(n2,displaylabels = TRUE)
#Removing Isolates
library(UserNetR)
data("ICTS_G10")
gden(ICTS_G10)
length(isolates(ICTS_G10))
n3 <- ICTS_G10
delete.vertices(n3,isolates(n3))
gden(n3)
length(isolates(n3))
#Filtering Based on Edge Values
data(DHHS)
d <- DHHS
gden(d)
op <- par(mar = rep(0,4))
gplot(d,gmode="graph",edge.lwd = d %e% 'collab',
edge.col = "grey50", vertex.col = "lightblue",
vertex.cex = 1.0, vertex.sides=20)
as.sociomatrix(d)[ 1:6,1:6]
list.edge.attributes(d)
as.sociomatrix(d,attrname="collab")[1:6,1:6]
table(d %e% "collab")
d.val <- as.sociomatrix(d,attrname="collab")
d.val[d.val < 3] <0
d.filt <- as.network(d.val, directed=FALSE,matrix.type="a",
ignore.eval=FALSE,names.eval="collab")
summary(d.filt, print.adj=FALSE)
gden(d.filt)
#Method to drawing
op <- par(mar = rep(0,4))
gplot(d.filt,gmode="graph",displaylabels = TRUE,vertex.col="lightblue",vertex.cex=1.3,
label.cex=0.4, label.pos=5,
displayisolates = FALSE)
#Advanced
op <- par(mar = rep(0,4))
d.value <- as.sociomatrix(d,attrnment="collab")
gplot(d.filt,gmode="graph",thresh=2,vertex.col ="lightblue",vertex.cex=1.3,
label.cex=0.4, label.pos=5,
displayisolates = FALSE)
par(op)
#Transformating a directed network to non-directed network
net1mat <- symmetrize(net1, rule = "weak")
net1symm <- network(net1mat,matrix.type="adjacency")
network.vertex.names(net1symm) <- c("A","B","C","D","E")
summary(net1symm)