論文閱讀:Detecting differentially expressed genes by smoothing effect of gene length on variance estimation
關於RNAseq Normaization更進一步閱讀
表現差異分析(Differentially Expression Analysis)是RNAseq實驗很重要的目的之一,因為我們想了解各種狀態間基因表現的差異!但目前大部分用來做DE analysis的演算法有一個問題,便是傾向於將較長的基因辨識為有表現差異的基因,而比較不容易將長度短的基因辨識為有表現差異的。
這篇研究認為這演算法的偏差,是來自於其計算過程中沒有將gene length對於reads count的variance estimation考慮進去,所以想提出一種改進過的方法LenSeq,將基因的長度smoothing,以達到讓有表現的短片段基因能被辨識出來!
原本科學家都認為RNAseq可以unbiased的用來比較基因的表現量,但實際上定序出來的reads是正比於此transcripts的長度,且定序深度也跟reads的總處相關,
換句話說,在資料前處理的normalization需要考慮進去這個bias:reads count 正比transcripts length,有人會借用microarrary data所常使用的quantile normalization,但其實這個問題在microarrary的設計中不會遇到,所以這標準化的分析並沒有考慮進去這偏差。
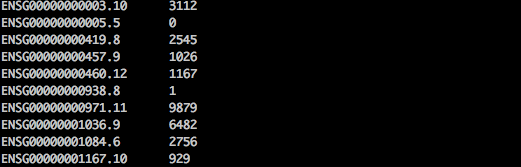
目前第一步用來表準化transcripts表現量的數值為RPKM(reads per kilo base per million mapped reads)=(read counts X 10^9)/( gene length X library size)或是後來的RPKM。可以用一個簡單的例子來思考:
|
|
Reads count for gene
|
Total Library Size
|
|
Library 1
|
800, 800, 800, 800, 800, 800, 800, 800, 800, 7800
|
15000
|
|
Library 2
|
1000, 1000, 1000, 1000, 1000, 1000, 1000, 1000, 1000, 6000
|
15000
|
當這兩個library size一樣的時候,會發現上面十個基因都是DE genes, 但實際上是不合理的,所以單純的使用library size表準化是不夠的。
後來的DESeq和edgeR,則有更進一步的方式來進行標準化,他們的假設建立在大多數的基因表現是沒有Deffierential Expression的,edgeR有工具可以trimmed mean of M-value,其會將read counts較高的數值去掉,剩下的再來計算兩個要比較的library之weighted mean of log ratio ,而DESeq則是使用scaling factor來標準化,先計算每個基因在不同lanes間的geometric means,取得每個reads count對應到其geometric means的ratio,在使用這群數值的mean 來代表scaling factor,最後在應用這數值來標準化兩個library。本質上這樣的演算法是將read counts過大或是過小的transcripts濾掉,從新調整資料的centrality。
小結論,仔細思考RNAseq的normalization步驟是很重要的,其影響到downstream analysis。
Bullard JH, Purdom E, Hansen KD, Dudoit S, Evaluation of statistical methods for normalization and differential expression in MRNA-Seq experiments, BMC Bioinfor- matics 11(1):94, 2010(被引用673次)
留一些關鍵字待查:
RNAseq followed Poisson distribution
Robinson MD, Smyth GK, Moderated statistical tests for assessing diFFerences in tag abundance, Bioinformatics 23(21):2881–2887, 2007.(被引用356次)
Negative binomial distribution
Whitaker L, On the Poisson law of small numbers, Biometrika 10(4):36–71, 1914(被引用65次)