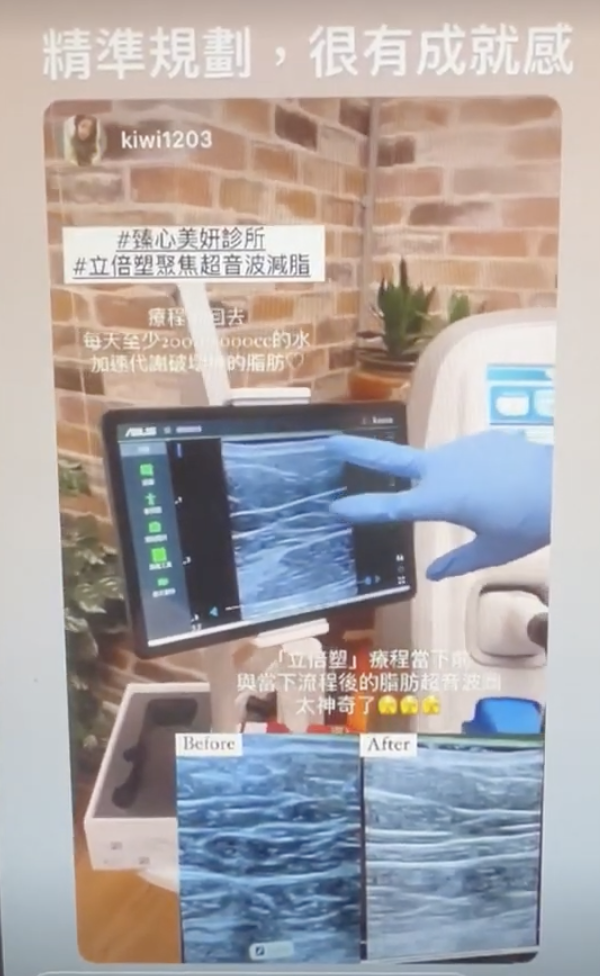
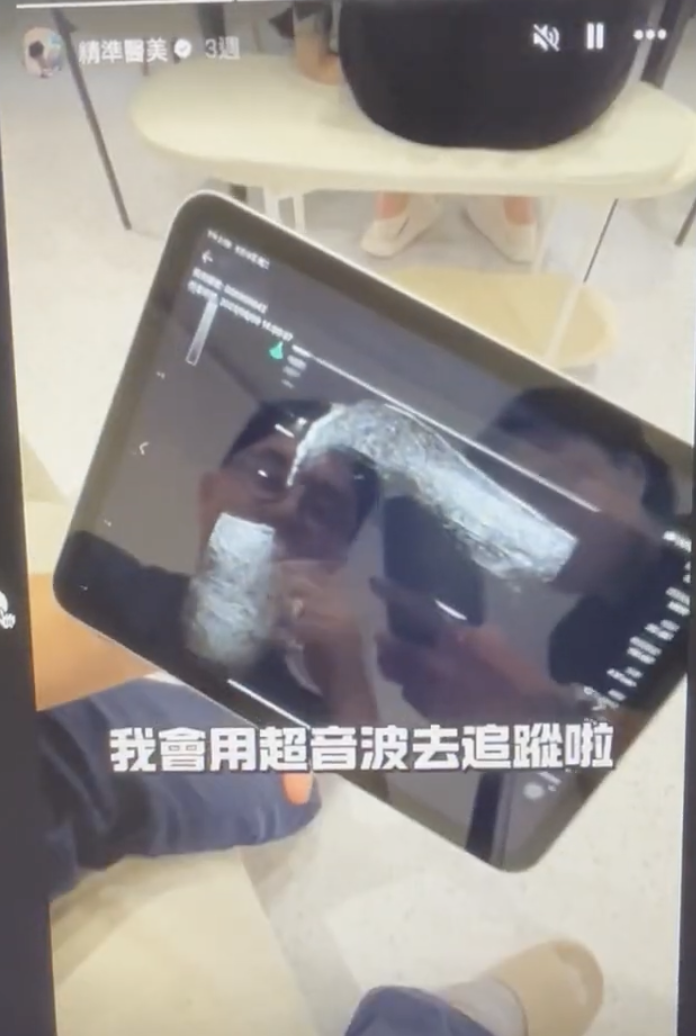
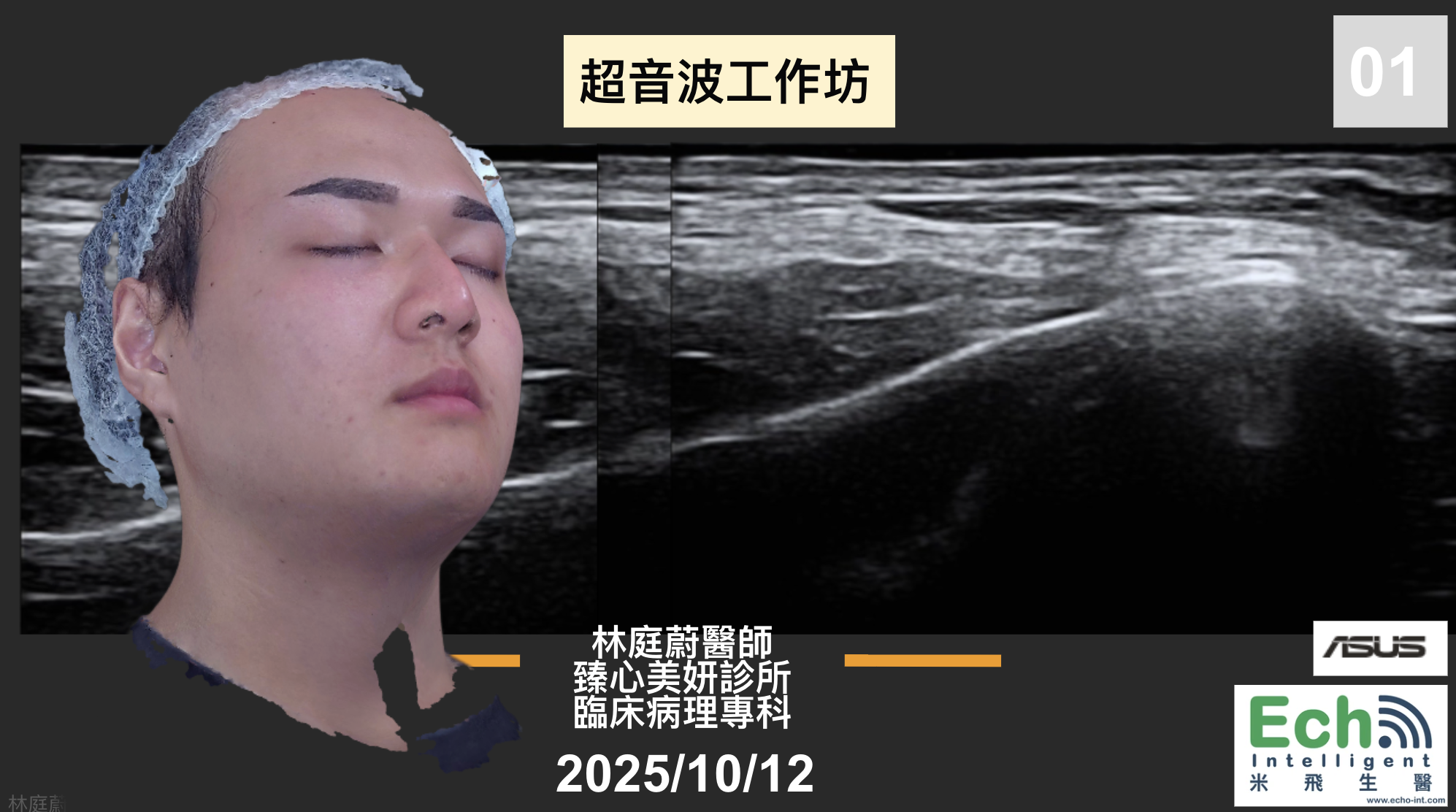
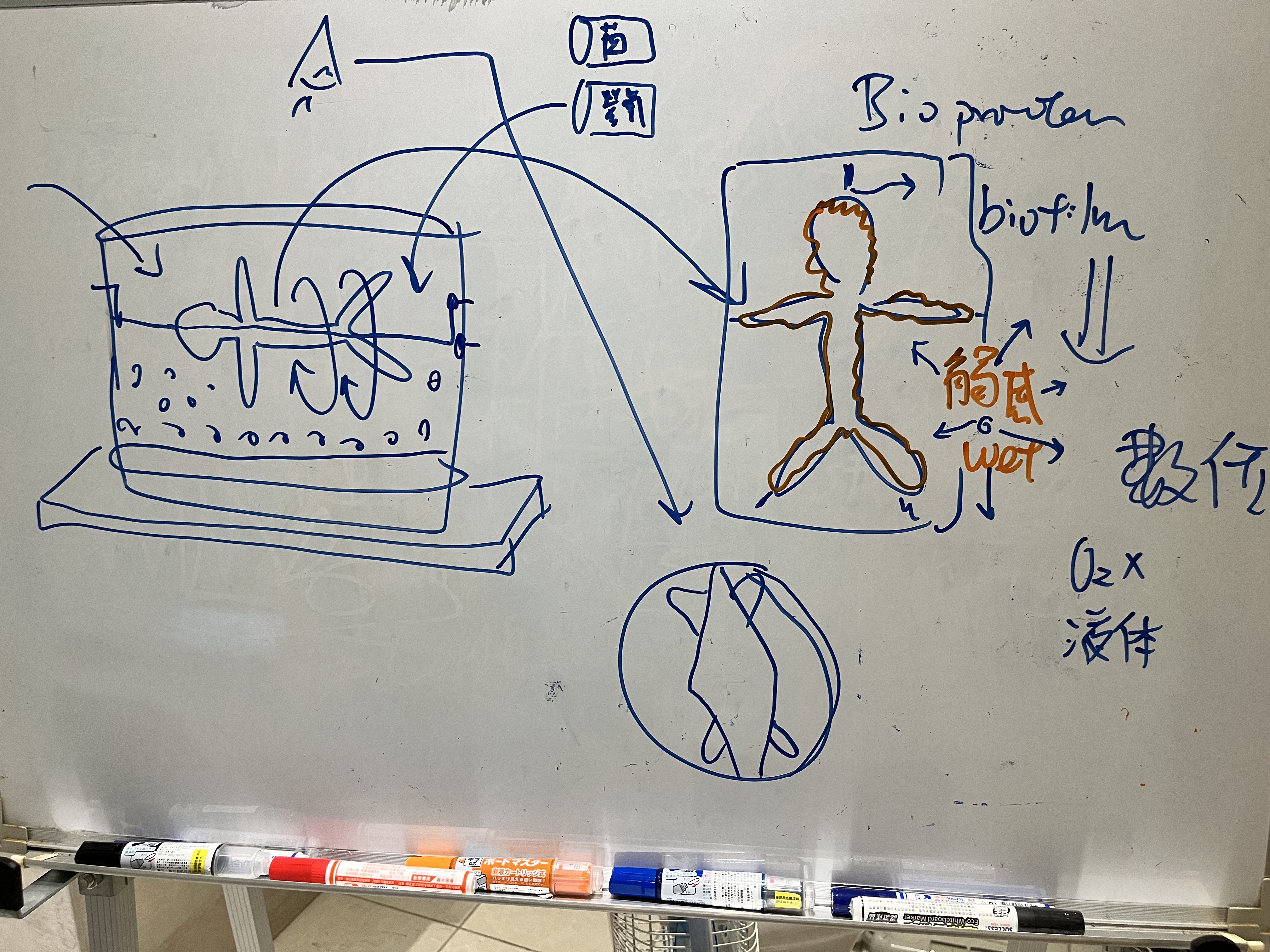
而去設計這樣的課程,知道最重要的是不要自己重新做輪子,所以發現目前國外超音波課程主要有Dr. Leonie Schelke和Dr. Peter Velthuis、Dr. Barbara Parda,他們有一套教學網站和實體課程,先從線上課程的方式去觀摩,他們的架構是比較偏向對解剖學結構的理解,當然一開始我也只能從這個角度來了解,不過慢慢地發現其實有更多的可能性,比如治療前的診斷工具或是規劃工具,這塊就增加很多能讓療程效果和安全更好的能力。
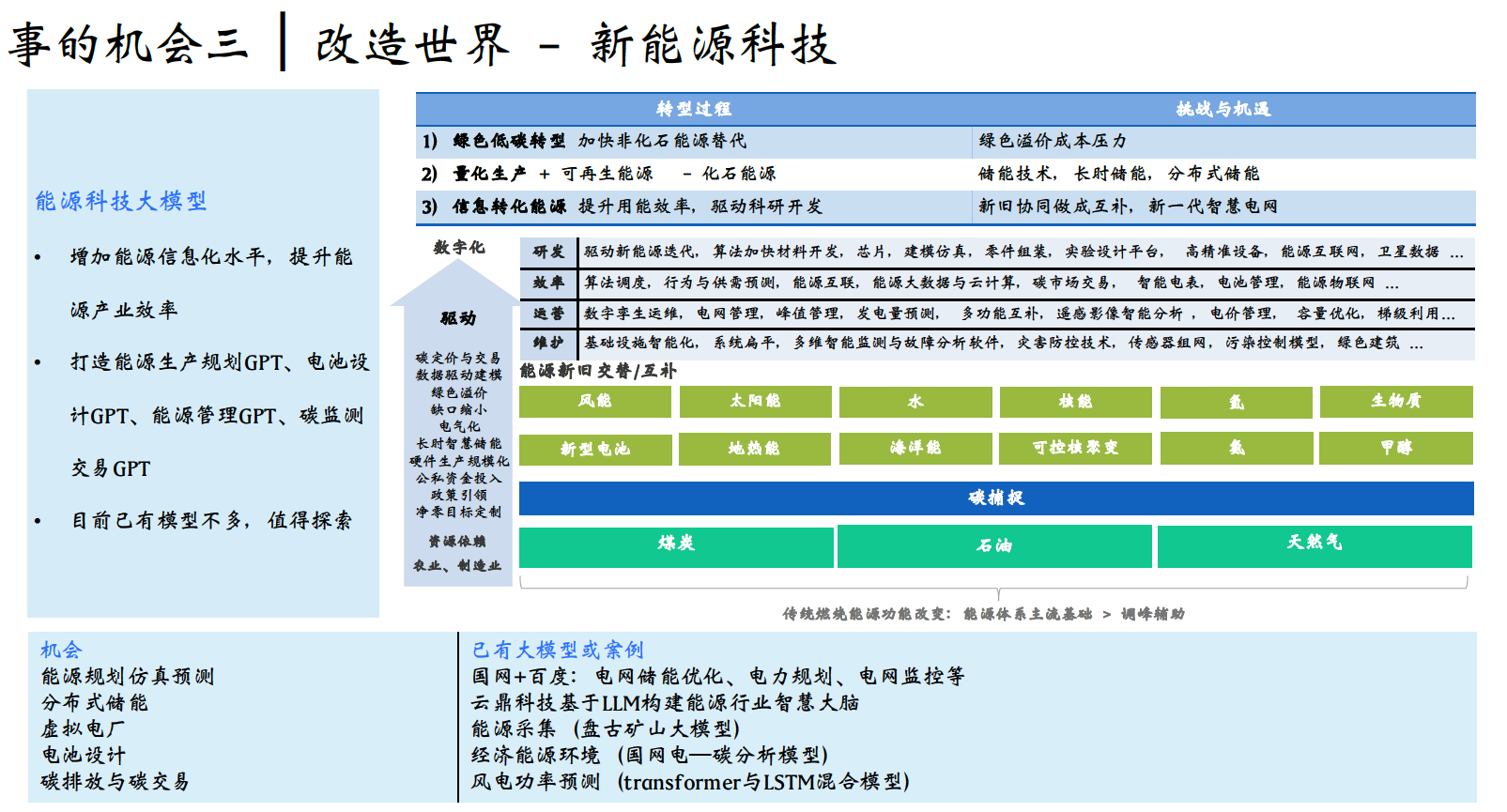
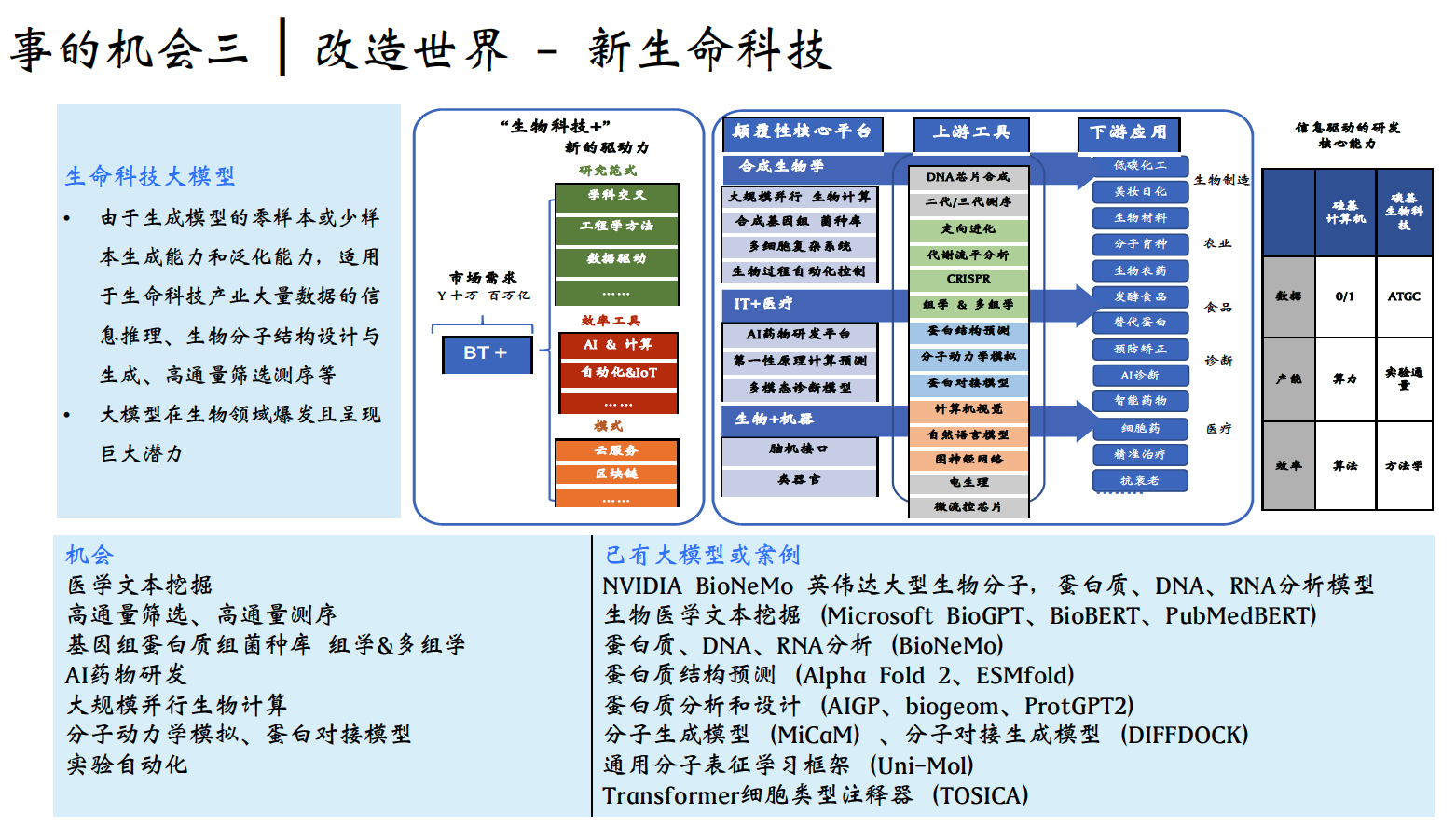
Kim HJ, Youn KH, Kim JS, Kim YS, Hong SO, Na J. Ultrasonographic Anatomy of the Face and Neck for Minimally Invasive Procedures: An Anatomic Guideline for Ultrasonography. 1st ed. Springer (Singapore); 2021. ISBN: 9789811565595.
Urdiales-Gálvez F, et al. Ultrasound in aesthetic dermatology (2021). J Cosmet Dermatol. 2021.
Ingallina F, et al. (2022).
超音波系列綜述 (Diagnostics 期刊)
Ultrasonography of the upper face. Diagnostics. 2024;14:1718.
Ultrasonography of the middle face. Diagnostics. 2024;14:2544.
Ultrasonography of the lower face. Diagnostics. 2025;15:921.
Is now really the time to start your hackerspace? Shouldn’t you wait?Have you really thought of all the problems?
Sure it it eh time!It’s always easier to ask forgiveness than it is to get permission — Grace Hopper It’s important to start. Many problems you think of before will banish as soon as you get start
基本軟價值的規劃,用哪些工具來紀錄在這空間發生的科學探索活動,以及為後續商業開源行為做準備,畢竟對這類型主題有興趣的人,絕對是利用這類網路相關的工具來探索和協作的。目前我自己的第一個主軸項目是From Zero to Hero: manipulation of plasmids,就是從質體放大、純化到修改的流程跑一次,中間希望可以使用到Ailurus,這間我其實關注超過三年的公司,所開發的蛋白質表達系統,其牽涉到的概念,所謂的condensate,蠻有趣的,值得藉此來多探索。
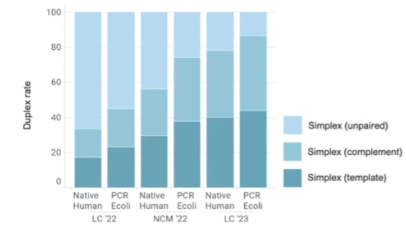
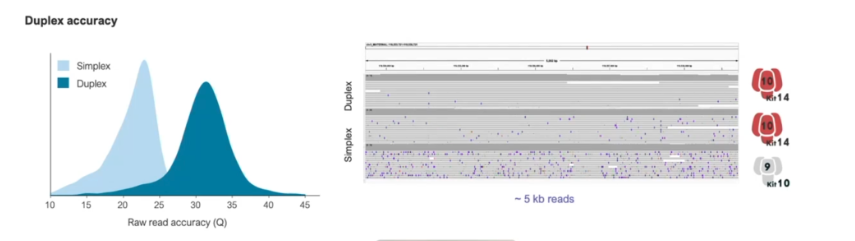
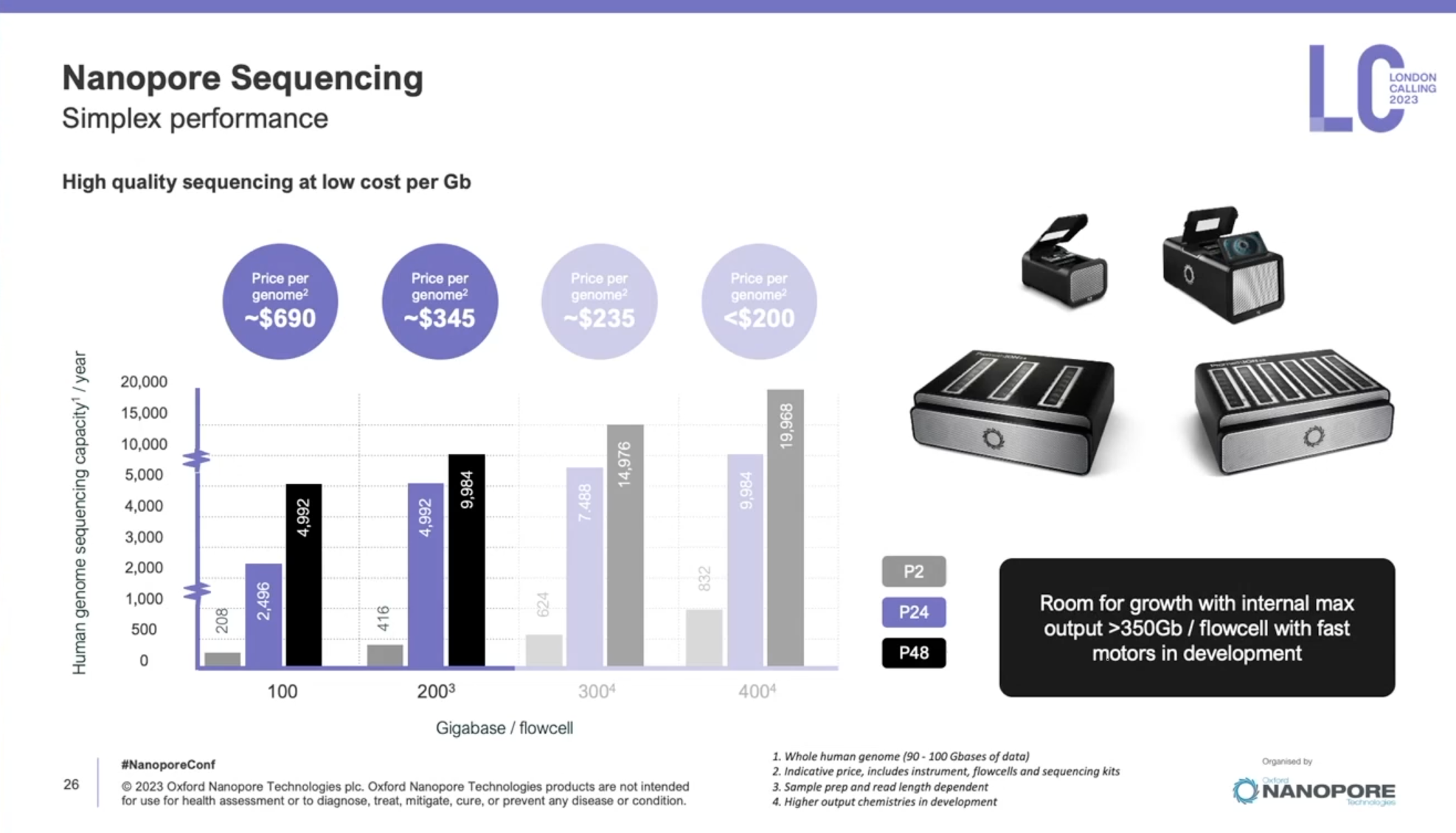
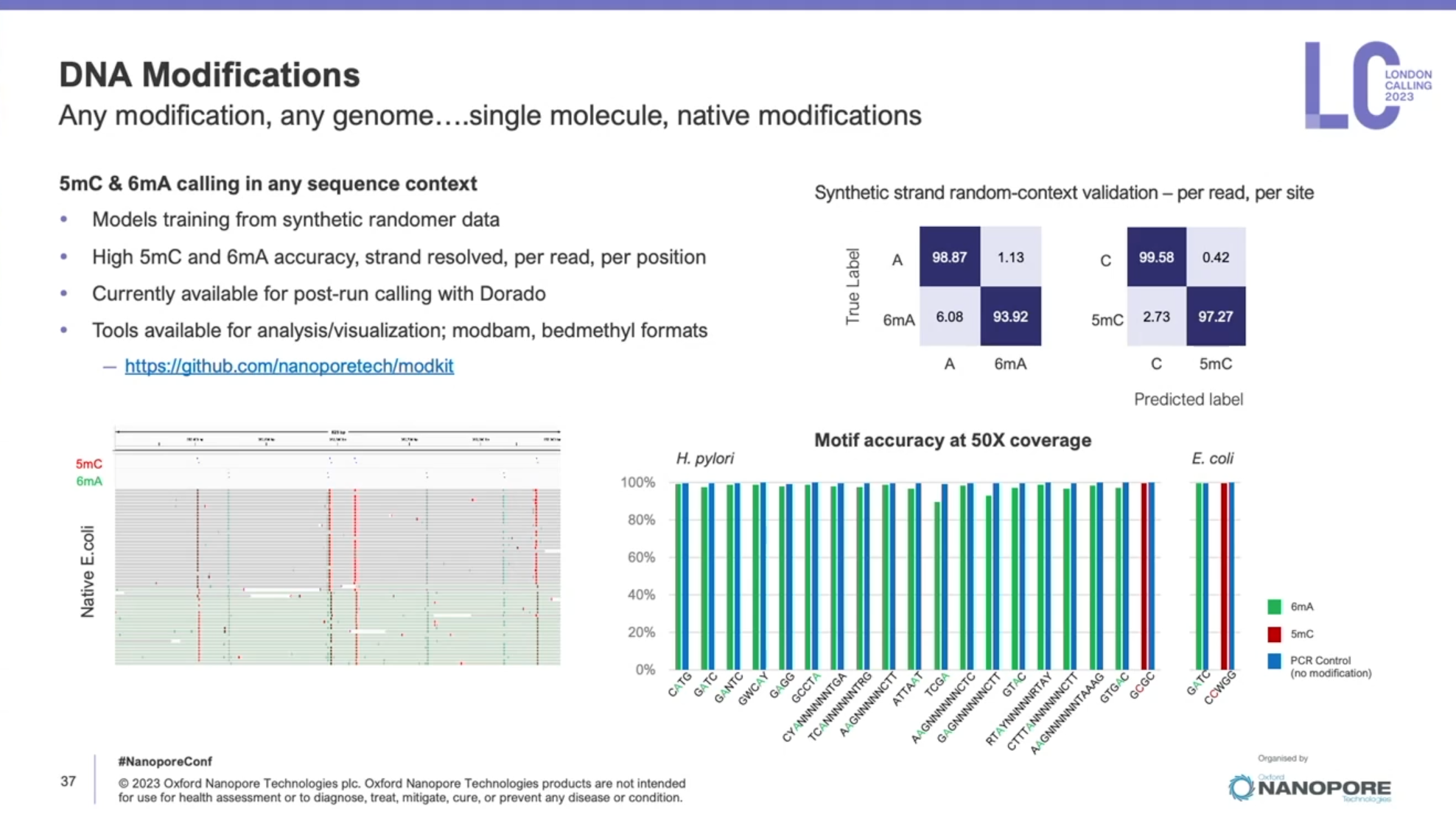
一年一度的Oxford nanopore大會又來了,假如時間不夠,可以直接聽牛津孔洞的CEO James Clarke的演講Update from Oxford Nanopore Technologies,基本上就會是今年他們火力集中的部分,大部分會議中的演講,會慢慢在他們官方的youtube頻道釋出。
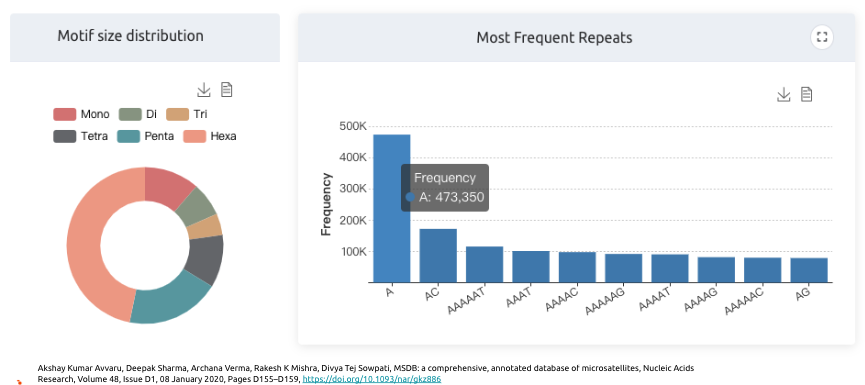
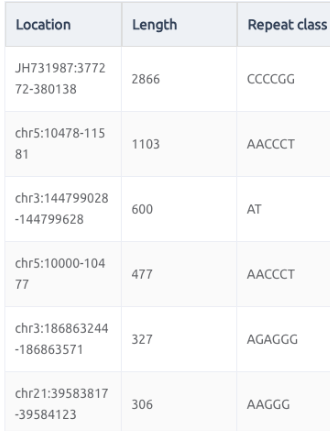
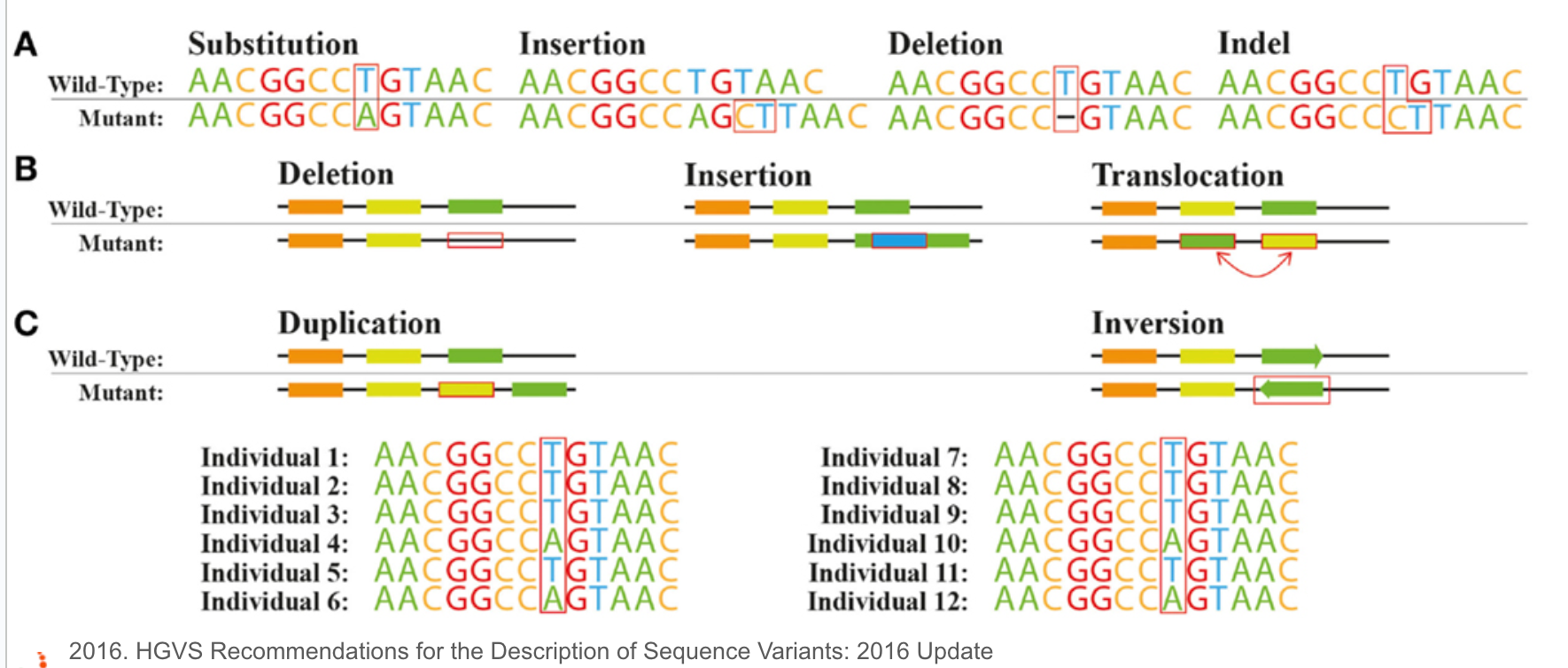
短串聯重複(Short Tandem Repeats,簡稱STRs)和微衛星(Microsatellites)這兩個在形容基因組結構的名詞,其實本質上是一樣的,但在體細胞談論遺傳時,都是以短串聯重複STRs為主,而在腫瘤領域在探討這類短片段重複的序列特性,則是在。它們都指的是一段由短的核苷酸序列(通常為2-6個核苷酸)構成的重複單元在基因組中連續出現的區域。
而這樣的重複片段在不同基因結構的區域,其實也有不少相關疾病被知道,比如上面圖片來自Hannan, A. J. (2018). Tandem repeats mediating genetic plasticity in health and disease. Nature Reviews Genetics, 19(5), 286-298.文章,便可以看到下面相關的疾病:
短串聯重複(Short Tandem Repeats,STRs)在基因組中具有高度變異性,與某些遺傳性疾病密切相關。以下是幾個與STRs相關的遺傳疾病範例:
肺動脈高壓(Pulmonary Arterial Hypertension,PAH):這是一種影響肺血管的疾病,與BMPR2基因中的STRs變異相關。該變異導致骨形成蛋白受體2(bone morphogenetic protein receptor type 2)的功能缺陷,使肺動脈壓力上升,進而引起心臟負擔加重。
拷貝數變異(Copy Number Variants):在長一點的重複序列
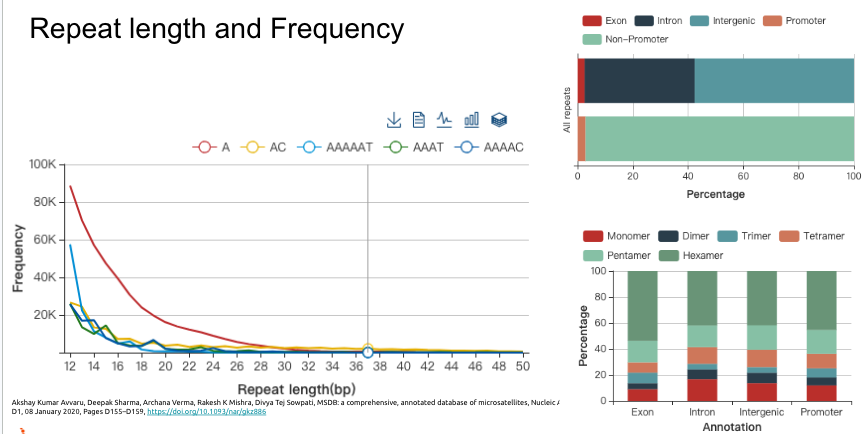
上面的短串聯重複片段(STRs),既然叫做“短”,那麼就有在長一點點的重複序列特徵,那麼就會被歸類在所謂的拷貝數變異,歸類在更大範圍的結構變化(Structual Variation),換句話說,拷貝數變異(Copy Number Variants)涵蓋了一個相對廣泛的基因組片段大小範,從數百個核苷酸(bp)到數百萬個核苷酸(bp)不等。這些變異可以是重複(增加拷貝數)或缺失(減少拷貝數)。然而,CNVs的精確大小範圍會根據定義和檢測方法而有所不同。一些研究將CNVs定義為影響至少1,000個核苷酸(1 kb)的變異,而其他研究則將閾值設置為50,000個核苷酸(50 kb)或更大。隨著檢測技術的不斷進步,研究人員現在能夠在更細的尺度上檢測到更小的CNVs,進一步擴大了我們對這些變異的認識和研究範疇。
Redon, R., Ishikawa, S., Fitch, K.R., Feuk, L., Perry, G.H., Andrews, T.D., Fiegler, H., Shapero, M.H., Carson, A.R., Chen, W., Cho, E.K., Dallaire, S., Freeman, J.L., Gonzalez, J.R., Gratacos, M., Huang, J., Kalaitzopoulos, D., Komura, D., MacDonald, J.R., Marshall, C.R., Mei, R., Montgomery, L., Nishimura, K., Okamura, K., Shen, F., Somerville, M.J., Tchinda, J., Valsesia, A., Woodwark, C., Yang, F., Zhang, J., Zerjal, T., Zhang, J., Armengol, L., Conrad, D.F., Estivill, X., Tyler-Smith, C., Carter, N.P., Aburatani, H., Lee, C., Jones, K.W., Scherer, S.W., & Hurles, M.E. (2006). “Global variation in copy number in the human genome." Nature, 444(7118), 444-454.
Stankiewicz, P., & Lupski, J.R. (2010). “Structural Variation in the Human Genome and its Role in Disease." Annual Review of Medicine, 61, 437-455.
Weischenfeldt, J., Symmons, O., Spitz, F., & Korbel, J.O. (2013). “Phenotypic Impact of Genomic Structural Variation: Insights from and for Human Disease." Nature Reviews Genetics, 14(2), 125-138.
Zarrei, M., MacDonald, J.R., Merico, D., & Scherer, S.W. (2015). “A Copy Number Variation Map of the Human Genome." Nature Reviews Genetics, 16(3), 172-183.
跟3D基因體學相關的文獻
Dekker, J., Marti-Renom, M. A., & Mirny, L. A. (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics, 14(6), 390-403.
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., … & Dekker, J. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326(5950), 289-293.
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., … & Ren, B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485(7398), 376-380.
Rao, S. S., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., … & Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665-1680.
Bonev, B., & Cavalli, G. (2016). Organization and function of the 3D genome. Nature Reviews Genetics, 17(11), 661-678.