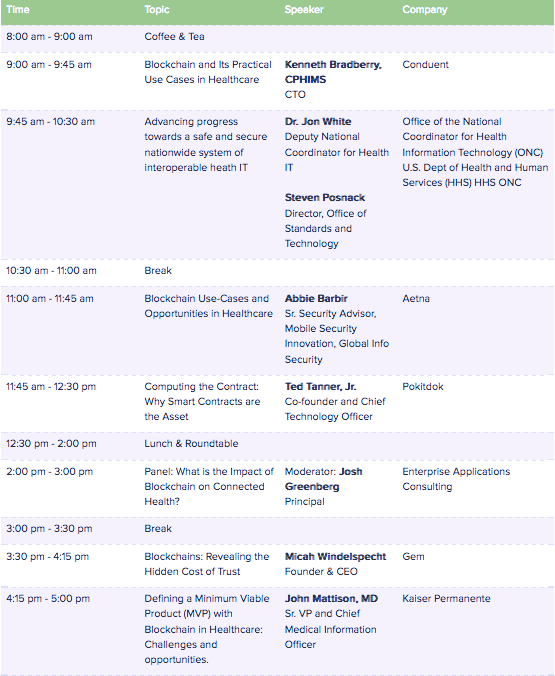
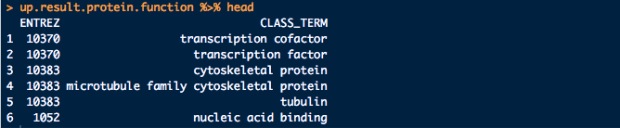
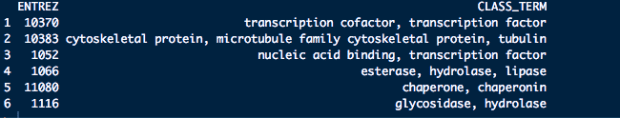
這邊嘗試要解決一個很棘手的問題,手上有一個下面這樣的資料清單,而每個資料夾下面有多個有固定編號的檔案。

而每筆資料大概長的如下面這樣:

而每個檔名是那筆資料的編號和資訊,最後希望可以把單個檔案讀取進來後,合併在一起,且根據每個檔案的檔名,新增4個欄位和其型態,最後理想上希望變成下面的樣子:
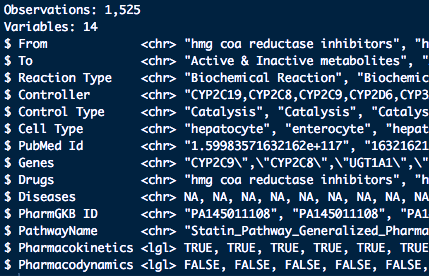
根據這個問題的話,可以使用read_delim, pmap_chr, map, pmap, reduce,這四個函數來使用,要完成這個小工作的話,能簡單可以分成幾個概念:
第一個是要彙整多個檔案路徑、第二個讀取多個檔案路徑的函數、第三個則是根據不同資料的檔名,來為其所讀取的data.frame新增欄位、第四個則是將所有放在list中的data.frame合併。
當概念解離清楚之後,便可以針對每一步挑選適用的函數來處理,比如
第一個小步驟想要彙整多個檔案路徑,這步要準備的便是把所有路徑以list的方式來儲存,然後用paste0函數來將每個路徑合起來,使用pmap_chr來完成這件事。
pmap_chr函數可以用函數來處理list中的每個element,接者在使用map搭配read_delim來完成第二個小步驟,如下面這個代碼
#將所有的變數,使用list來儲存
list(as.list(rep(Pathway.path, pathway.number)),as.list(rep("/",pathway.number)),as.list(list.pathway.json)) %>%
#將這些變數導入到pmap_chr函數,因為pmap_chr是當list內的element為chr時所用的,這一部是為了把每個檔案的路徑組合起來
pmap_chr(paste0) %>%
#接者在使用map函數來針對上面處理完的一串路徑list,使用read_delim函數,分別讀取,並且在用list來吐出
map(read_delim,delim="\t")
第三個則是根據不同的資料檔名,來為其所讀取的data.frame新增欄位
此時我們已取得一個list,裡面的element都是一個檔案讀入的data.frame,而我們希望再把data.frame合併前,將每個data.frame使用其檔案名稱來增添其資料欄位,如下面的代碼:
#這邊的.代表從剛剛上面的代碼pipe進來的位置,後面三個則是我們希望增加的欄位,和每個欄位的名稱
list(.,pharmGKB_ID,pathwayName,Pharmacokinetics,Pharmacodynamics) %>%
#同樣的這邊可以使用pmap,此時不同使用pmap_chr,因為我們list中,一組是data.frame的格式
pmap(function(x,pharmGKB_ID,pathwayName,pharmacokinetics,pharmacodynamics){
x %>%
mutate(`PharmGKB ID`=pharmGKB_ID,
`PathwayName`=pathwayName,
Pharmacokinetics=pharmacokinetics,
Pharmacodynamics=pharmacodynamics )
})
#這邊比較值得一提的是pmap裡面可以使用函數作為參數,而這個函數可以直接在裡面定義,有點套用lambda的寫法,來做這件事情,這邊是為了把欄位的名稱確立
第四個則是將所有放在list中的data.frame合併,這邊使用reduce來解決這件事情,reduce可以將放入的list,把每個element用某個函數處理,一個接一個。
CleanPathway <-list(as.list(rep(Pathway.path, pathway.number)), as.list(rep("/", pathway.number)),as.list(list.pathway.json)) %>%
pmap_chr(paste0) %>%
map(read_delim,delim="\t") %>%
list(.,pharmGKB_ID,pathwayName,Pharmacokinetics,Pharmacodynamics) %>%
pmap(function(x,pharmGKB_ID,pathwayName,pharmacokinetics,pharmacodynamics){
x %>% mutate(`PharmGKB ID`=pharmGKB_ID,
`PathwayName`=pathwayName,
Pharmacokinetics=pharmacokinetics,
Pharmacodynamics=pharmacodynamics )
}) %>%
reduce(rbind)
這四個步驟便可以完成這件事情,這過程很重要的便是搞清楚map, pmap, reduce,他們要怎麼接受參數和處理,可以因此更清楚在R裡面list和data.frame的特性。不過在處理的過程也會使用到stringr的函數,來處理字串的問題。
閱讀參考:
How can I read in multiple files
Reading and combining many tidy data files into R