目前ggplot2基於grid建立了個龐大的繪圖系統,因此當然會需要使用oop來管理日漸複雜的ggplot2 “world",在ggplot2 2.0.0 版提供了官方的文檔在於ggplot2底層的資訊,在Wickham Hadley一開始寫ggplot時,沒有預想到如今如此廣泛的使用和許多套件及開發者的參與,所以在底層物件撰寫上有許多可改進的地方,以幫助跨套件的移植性和讓開發者能使用ggplot2裡面的基礎往上做出更豐富的視覺化。
在ggplot2 2.0.0釋出了ggproto的詳細文檔,而ggproto是基於proto這個oop系統修改而成的“微”面向編成,具有跨套件的繼承性(cross-package inheritance),另外,因為ggplot2為基於grib的繪圖系統,所以到這邊開始需要基礎的grid知識,會比較能融會貫通兩者的關聯性,這邊先專注解釋ggproto裡面的架構,之後會在grid的部分詳加說明。
這部分可能先對R中的OOP系統多少理解一下,會比較能理解他們語法的初衷,可參考前一篇文章
建立ggproto物件
| # Construct new_Geom class inheired from Geom class | |
| new_Geom <- ggproto("new_Geom", Geom, | |
| required_aes = c("x", "y"), | |
| defaut_aes = aes(fill="yello"), | |
| draw_key_key = draw_key_polygon, | |
| draw_group = function(data, panel_scales, coord){ | |
| coords <- coord$transform(data, panel_scales) | |
| grid::polygonGrob( | |
| x = coords_df$x, | |
| y = coords_df$y, | |
| gp = grid::gpar(col = coords_df$colour, | |
| fill = coords_df$fill, | |
| alpha = coords_df$alpha) | |
| ) | |
| } |
| # create ggproto object | |
| # 會發現語法承襲自proto的系統,不需要像是RC system的field等 | |
| new_ggproto_object <- ggproto("Class_name", | |
| parent_ggproto_class, | |
| x = 0 | |
| f1_method = function(self,n){ | |
| self$x <- self$x + n | |
| self$x | |
| } | |
目前已有ggplot2::Geom, ggplot2::Stat, ggplot2::Position, ggplot2::Scale這四個內建的ggproto類別,而在做延伸ggplot2視覺化的函數時可以用上面四個類別來起手。
Geom類別
– draw_panel(self, data, panel_scales, coord) or draw_group(self, data, panel_scales, coord)
– required_aes
– default_aes
Coord類別
– aspect
– labels
– render_bg
– render_fg
– render_axis_h
– render_axis_v
– range
– train
– transform
– distance
– is_linear
Facet類別
– compute_layer
– map_data
_ draw_panel
Stat類別
– finish_layer
– setup_params
– setup_data
– required_aws
– default_area














 Illumina sequence identifiers
Illumina sequence identifiers

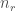 代表單一個gene其上的reads數量,L代表是這段基因的總長度除1000(轉換成kilobase的單位),N則是總共的reads數,通常用於singel reads
代表單一個gene其上的reads數量,L代表是這段基因的總長度除1000(轉換成kilobase的單位),N則是總共的reads數,通常用於singel reads


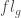 則代表是這基因的exon總長度
則代表是這基因的exon總長度