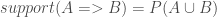Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nature Reviews Cancer, 16(9), 582–598. http://doi.org/10.1038/nrc.2016.73

這篇文獻回顧是關於纖維母細胞(Fibroblast)在癌症中角色,近年來對於癌症細胞所處的微環境在癌細胞癌化和轉移中扮演越來越重要的角色,其中纖維母細胞便是要角。
撰寫者為Raghu Kalluri教授,目前是MD Anderson Cancer Center擔任癌症生物研究中心(Cancer Biology Department)和癌症轉移研究中的負責人(Metastasis Research Center),本身是MD/PhD,其研究關注癌症組織環境對於癌症細胞的惡化和轉移的影響。這裡有Raghu Kalluri教授再冷泉港會議中接受訪談的影片,裡頭有其對於其研究室近年研究方向的想法。
微環境之於癌症細胞的重要性
關於身體對於癌組織的反應,對於最終腫瘤組成很重要。這些癌組織微環境還被稱為Desmoplastic reaction, tumour stroma, tumour microenvironment(TME)。像是微環境中的免疫細胞、angiogenesis、oxygen tension、interstitual pressure, ECM remodeling和代謝產物的改變都是近年來認為跟癌細胞發展和轉移重要的因素。其中,這些癌組織的周圍細胞中,纖維母細胞是最為重要的一個角色。這些跟癌症有關的纖維母細胞被稱作Cancer-associated fibroblast。
此篇文獻回顧內容大致可以分成這幾個要點:
1. 關於纖維母細胞研究的起源
2. 激化的纖維母細胞和未激化的纖維母細胞的差異
3. 纖維母細胞跟生體內相關生理和病理上的角色:傷口癒合,組織纖維化
4. 跟癌症的連結:所謂的癌症組織的纖維化現象(Cancer Fibrobsis)
5. 激化纖維母細胞的多樣性(來源、組織特異性、狀態特異性)
6. 表觀遺傳調控在纖維母細胞的表型影響
7. 癌症相關的纖維母細胞
a. 對於癌化現象的角色
b. 在代謝中扮演的角色
c. 在免疫中扮演的角色
d. 對於抗藥性的關係
關於纖維母細胞的活化
通常纖維細胞在組織內的狀態是“未激活”(quiescent, resting),在組織受傷的時候才會被激活,而這些狀態還可以分為reversible 或是irreversible兩種,可能由epigenetic modification造成的。所謂的活化狀態的纖維母細胞最一開始的定義是由ACTA2活化。這個基因剛好是調控體內平滑肌的。而這類型的活化纖維母細胞為疤和慢性傷口的主要參與纖維母細胞。而纖維母細胞對於環境壓力的適應能力,讓其被視為在癌組受損時,能參與癌症復發的角色。
The host response to evolving cancer cells results in the generation of tumour tissue that contains components of normal organs. Such robust host responses define complex heterogeneity interactions of cancer tissue with host cells and are known as desmoplastic reaction, tumour stroma or tumour microenvironment.
纖維母細胞研究起源:
1858年在顯微鏡下,對於那些在微血管旁邊,沒有跟basement membrane緊貼,不是免疫細胞,也不是上皮細胞,可能是結締組織的一部份稱作纖維母細胞. 在一開始認為纖維母細胞在一般情況下,對於組織內的代謝、各種表現不太有影響力,唯有在特定情況下才會激活,如受傷後急性發炎和慢性發炎以及組織纖維化的過程。
比較精準的定義,應該稱纖維母細胞為resting mesenchymal cell,且可以被激活成mesenchymal stem cell.這些細胞在TGFbeta, PDGF, IL-6刺激下,可以快速proliferate. 所以其實大多狀況下,我們所分離出來的纖維母細胞其實是就是所謂“活化”的,他們可以synthesizing ECM, 分泌cytokines, chemokines, 招集immune cell, exerting physical forces to modify tissue architecture。這樣的特性,在一些研究中被認為會加速癌細胞的轉移能力,其中有研究顯示BM-MSC會加速breast-cancer cell的轉移能力。
纖維母細胞在病理狀況下的角色
急性的傷口癒合
“any injuries to the functional parenchyma results in a host response. Injurious stimuli(mechanical trauma, radiation-induced or extreme temperature-induced damage, toxins, pathogenes or metabolic impairments.)”
標準的傷口復原反應會招集免疫、發炎細胞來促進血管新生和ECM deposition. ECM中的type I, type III, type IV, and type V collagenes, laminins, fibronectin都是由被活化的纖維母細胞所產生的。另一方面,纖維母細胞也是這些ECM-degrading protease的來源(如Matrix metalloproteinase, underscoring their crucila role in maintaing ECM homestasis by reguulation of ECM turnover).當傷口癒合後,活化的纖維母細胞會逐漸轉成不活化的狀態。
組織纖維化
當對組織的傷害是持續性的,這類傷害可以是慢性物理性,毒素, 代謝改變或是自體免疫.這樣持續性的修復反應便會造成所謂的纖維化.這過程主要是由表觀遺傳學所調控,造成持續激活狀態的纖維母細胞,且活化 anti-apoptosis pathway and initiaitng proliferation. (51-54)
癌症纖維化(cancer fibrosis)
癌細胞就像是永遠不會癒合的傷口,所謂的cancer fibrosis又稱做desmoplastic reaction, tumour stroma and TME。目前已知道myofibroblast在癌症惡化和轉移有所關聯,但目前發現他的功能是很bimodal的。招集組織間纖維母細胞到癌症細胞周圍的,除了growth factor外,還有免疫細胞。在細胞損傷中,TGFbeta, PDGF, fibroblast growth factor 2(FGF2)都佔有很重要的角色。在很多癌症裡面,TGFbeta在招募活化的纖維母細胞都具有重要角色,且局部的癌症相關纖維母細胞(Cancer-associated Fibroblast)活化可能也是由在TME中的TGFbeta很有關係。
有研究顯示,在早期的癌化過程中,免疫細胞分泌的IL-1beta能活化纖維母細胞中的nNF-kB的訊息路徑,產生癌化前期的secretome。甚至有研究在具有癌症體質的成年公雞上,製造傷口,會使得傷口組織產生有侵襲性的癌症。在正常器官中,活化的纖維母細胞數量不多,但在癌症組織中,卻具有相當大量的活化纖維母細胞在組織之中,由組織裡上升的alphaSMA和FAP當作證據。在組織招募新的微血管和累積免疫細胞於癌症細胞的環境中,這些生長因子佔有非常重要的角色,如分泌VEGFA, PDGF, EGF,IL-6,IL-8。VEGF本身可以造成血管通透性增加,使得腫瘤富含ECM蛋白質,像是type I collagene, fibronectin等,可以刺激血管新生。隨者腫瘤長大,其會大量增加各種collagem, laminins, firbronectin, preoteoglycans, perostin and tenascin C。像是perostin/tenascin本來是不太會出現在正常現體組織,卻在乳癌中大量出現。換句話說,纖維母細胞催化癌化的過程,可能就是一種對於體內組織慢性傷口的反應。
纖維母細胞的功能和多樣性
現在已經知道有多個標誌可以用來觀察活化狀態的纖維母細胞,像是FSP1, S100A4, vimentin, alphaSMA, fap, pdgf RECEOTR-alpha, DDR-2。不同特異表現的纖維母細胞對於癌細胞的調控,也會有很大的差異,像是在乳癌研究中,缺少Caveolin 1表現的纖維母細胞,本身會對於癌症代謝上帶來調控(98,99),然後,高Caveolin 1表現的纖維母細胞也可以促進癌細胞的ECM modeling。作者也表示,這些活化纖維母細胞的特性可能也跟纖維母細胞原本組織來源相關,比如是從Marrow-derived precursors, endotheia cells, liver, pancreas stellate cells, resting tissue fibroblast。
纖維母細胞的表觀遺傳學調控
目前已知跟纖維母細胞調控癌細胞的相關訊息傳導路徑可能很多個(TCFbeta(18), 20),很多認為這些讓纖維母細胞活化,也可能跟表觀調控相關。
在Fibrobrosis-associated fibroblast(CAF)的研究中,發現使用5-azacytidine(一種demethylation藥物)可以使得FAFs的proliferate rate 下降,產生的collagen I 減少,alphaSMA減少。如活化JAK1,STAT3可已調控LIF,且同時抑制dnmt/jak活性,可以讓CAF轉換成具有invasive的能力。
癌症相關的纖維母細胞(CAFs)
在催生癌化上的角色
本質上在癌組織中取得的纖維母細胞的特質就不太一樣,其展現的移動能力(migratory ability),自泌生長因子(autocrine growth factor),chemokine的能力都非常強。這些可能是表觀遺傳上的調控所造成的。下面分別是一些證據,在反應癌症相關纖維母細胞對於組織癌化所扮演的角色
1. CAFs可以催化non-invasion的癌細胞表現invasion的能力
2. CAFs藉由分泌SDF-1(Stromal cell-derived factor 1/CXCL12)來催生血管新生
3. CAFs上升的HSF1(heat shock factor 1)和癌細胞的HSF-1 drivedn tumorigenic programme相關
4. Yes-associated protein 1(YAP-1)會造成ECM stiffening
5. CAFs產生如MMPs(ECM degrading protease)讓癌細胞較高的motility/invasion ability
6. CAFs缺少分泌TIMPs,加強分析MMPs和DISINTEGRIN,adam10,這些跟刺激癌細胞的幹細胞特性相關
7. 在肺癌中,癌細胞分泌THBS2可以活化Fibroblast,如此造成轉移的能力
ECM remodeling by fibroblast may participate in the generation and maintenance of the cancer stem cell niche. Perostin(POSTN)可能就是其中的關鍵,對於Wnt signalling-mediated cancer stem cell niche maintenance. Elevated WNT signaling is found in colon cancer stem cells that are proximal to CAFs
p.s
這篇文獻回顧跟癌細胞會剝離原本的ECM造成轉移這主題相關
Buchheit, C. L., Weigel, K. J., & Schafer, Z. T. (2014). Cancer cell survival during detachment from the ECM : multiple barriers to tumour progression. Nature Publishing Group, 14(9), 632–641. http://doi.org/10.1038/nrc3789
Zachary T. Schafer 研究這個主題的教授(ECM, anoikis-resistance)
在癌症細胞相關代謝
在CAFs內的代謝情況就跟highly proliferating的細胞一樣,在能量產生上很大程度依賴glycolysis,導致這些代謝調控的改變,可能是由於如TGFbeta, PDGF, hypoxia, hypoxia-inducible factor 1 alpha, ROS-mediated suppression of CAV1所造成的。這樣的現象(Warburg effect)同時伴隨者跟增加的catabolic activity/autophagy有關(TGF-beta signaling,IDH3 down-regulation)。CAF可能扮演重要的anabolic/catabolic balance in the regionally diverse milieu(環境)。Tryptophan/arginine starvation對於T cell活化和淋巴球的功能具有重要角色,CAFs藉由調控代謝環境,藉此對於免疫細胞的反應做出調控。
這篇論文在談論為何這些高度proliferate cell換依賴glycolysis呢?
Heiden, M. G. Vander, Cantley, L. C., Thompson, C. B., Mammalian, P., Exhibit, C., & Metabolism, A. (2009). Understanding the Warburg Effect : Cell Proliferation. Science, 324(May), 1029–1034. http://doi.org/10.1126/science.1160809
這篇論文在談論癌症細胞在能量代謝平衡的問題
Fiaschi, T., Marini, A., Giannoni, E., Taddei, M. L., Gandellini, P., De Donatis, A., … Chiarugi, P. (2012). Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Research, 72(19), 5130–5140. http://doi.org/10.1158/0008-5472.CAN-12-1949
這篇文章有講到CAF/CSC裡 MCT1 increased, GLUT1 decreased
“Oncogenes induce the cancer-associated fibroblast phenotype: Metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery
Catabolic fibroblasts donate the necessary fuels (such as L-lactate, ketones, glutamine, other amino acids, and fatty acids) to anabolic cancer cells, to metabolize via their TCA cycle and oxidative phosphorylation (OXPHOS).Thus, oncogene activation (RAS, NFkB, TGF-β) and/or tumor suppressor loss (BRCA1) have similar functional effects on adjacent stromal fibroblasts, initiating “metabolic symbiosis” and the cancer-associated fibroblast phenotype.Targeting “fibroblast addiction” in primary and metastatic tumor cells may expose a critical Achilles’ heel, leading to disease regression in both sporadic and familial cancers.”
在癌症組織中纖維母細胞與免疫的關聯
CAF對於免疫功能的調控(pleiotropic immunomodulatory functions)主要是由他的secretome所主導的,分泌ECM protein, ECM-remodeling enzymes, production of a plethora of cytokines, chemokines. 目前的研究主要是展現了fibroblast secretome的heterogeneity,its differential impact on tumour biologt.未來可能需要更多fibroblast-specific deletion cytokine/chemokines in preclinical tumour models的實驗,可能才可以提供比較精準的模型。
一般來說,認為CAFs可以提供immunosuppressive TME.但整體還是要看在哪類的TME中。目前認為跟CAFs相關的secretome裡有IL-6, IL-4, IL-8, IL-10, TNF, TGFbeta, C-C motif chemokine ligand 2(CCL2), CCL5(RANTES),CXCL9, CXCL10,SDF1, PGE2, NO, HGF, Human Leukocyte Antigen G(HLAG)。
IL-6 signaling被認為可能會抑制DCs的成熟,讓T cell無法活化,造成T cell anergy,另外,IL-6 可以使得monocyte differentiate into macrophage lineage和活化mast cells.但還不清楚是單純IL-6就可以,還是要其他的cytokine參與,其中TGFbeta也被認為在其中扮演重要的角色,可能和其調控Thelp 17 cell相關。
CXCL14從CAF而來,也被認為可以造成macrophage recrute到腫瘤。
CCL2 in breast cancer 則認為跟這件事有關,且其同時會被macrophage,DCs所分泌。
T cell recruitment所需的cyokine在CAF的secretome也可以找到(CXCL9, CXCL10, SDF-1),同時像是negative co-regulatory immune signal(PDL-1/2)跟抑制T cell activation有關。
targeting FAP+ CAF顯示出anitumour的效果,其藉由intramoural recruitmnet of CD+8 T cell SC04 cd+8 cell-mediated cancer cell killing
癌症相關纖維母細胞和抗藥性的關係
“mechanisms of resistance involving the stroma include the modulation of pathways involving the stroma include the modulation of pathways involving cacner cell-ECM/ CAF-ECM adhesion, cytokine, chemokine…。"
下面這三篇都在談論TEM對於藥物耐受性的關係
Correia, A. L., & Bissell, M. J. (2012). The tumor microenvironment is a dominant force in multidrug resistance. Drug Resistance Updates, 15(1–2), 39–49. http://doi.org/10.1016/j.drup.2012.01.006
Dittmer, J., & Leyh, B. (2015). The impact of tumor stroma on drug response in breast cancer. Seminars in Cancer Biology, 31, 3–15. http://doi.org/10.1016/j.semcancer.2014.05.006
Ohlund, D., Elyada, E., & Tuveson, D. (2014). Fibroblast heterogeneity in the cancer wound. Journal of Experimental Medicine, 211(8), 1503–1523. http://doi.org/10.1084/jem.20140692
CAF-mediated immune modulation, pro-angiogenic actions and metabolic reprogramming of the TME might aid in cancer cell survival and facilitate escape from therapy-induced cancer control。
 Cytoscape是一款開源免費的網絡視覺化和分析工具,專注在處理生物分子、蛋白質交互網絡,對於生物資料庫的整合頗佳,且其最有名的就是許多插件可以安裝,近年來在3.0版(2013)之後,Cytoscape軟件整個架構大改版,更模組化,且具有Restful API,對於一個從2001便開始的專案,真的是很努力在進步。
Cytoscape是一款開源免費的網絡視覺化和分析工具,專注在處理生物分子、蛋白質交互網絡,對於生物資料庫的整合頗佳,且其最有名的就是許多插件可以安裝,近年來在3.0版(2013)之後,Cytoscape軟件整個架構大改版,更模組化,且具有Restful API,對於一個從2001便開始的專案,真的是很努力在進步。

![computer=>antivirus\_software\[support=60%,confidence=60%\]](https://s0.wp.com/latex.php?latex=computer%3D%3Eantivirus%5C_software%5C%5Bsupport%3D60%25%2Cconfidence%3D60%25%5C%5D+&bg=ffffff&fg=404040%3B&s=0&c=20201002)