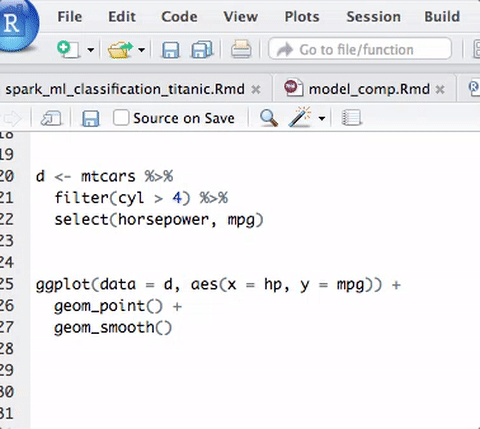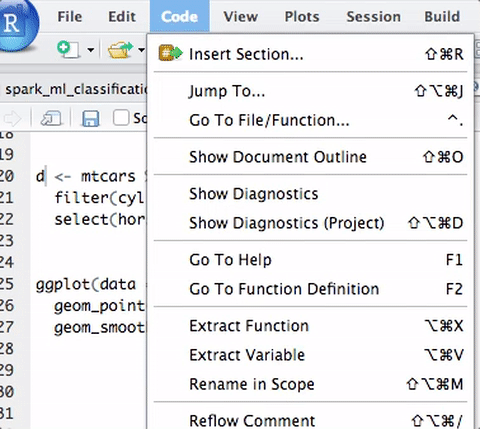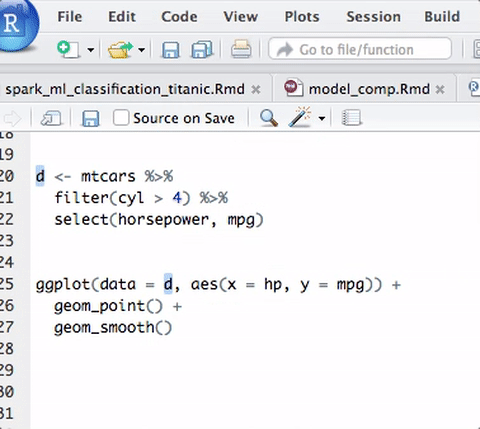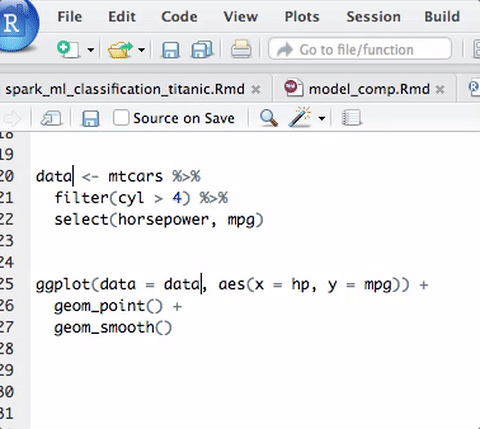
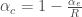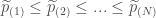purrr package 是Hadley Wickham和Lionel Henry所開發的,補強R base中原本的fucntional programming tool,帶入其他語言的特性,整體的風格是使用underscore.js, lodash及 lazy.js的語法。
R base語法中本身就有很多函數帶有部分Functional Programming意味的風格,如any,which,do.call,apply, sapply, vapply,lapply等等,只是沒有好好的統一,Hadley Wickham恰好把這部分補足了, 且裡頭的函數都是所謂的type-stable pure function導向,正如其package的名稱purrrr。
functional programming 其實是奠基在lambda calulus的觀念上,專注在四個基石上:資料(numbers, strings)、變數(函數的參數)、函數、函數應用(如何用函數修飾函數),概念上有點像是把Object-oriented programming中,function變成一個object,且專注於function功能的實現,用function去處理另一個function,去減少loop傳統寫法的使用,和重複性argument使用的麻煩。
基本上,purrr package中的函數便是依照許多其他functional programming導向的語言如scala、erlang、lisp等等特性來做的,簡單有這幾類的函數:
| Category | Related Function |
|---|---|
| Map | map(), map_chr(), map_int(), map_dbl(), map_df(), walk() |
| Reduce | reduce(), reduce_right() |
| Search | contains(), detect(), detect_index() |
| Filter | keep(), discrad(), every(), some() |
而觀念上的一些使用如recursion寫法、expression的應用等,都可以簡化和提高代碼的效果。
這整個項目的特性:
1. 將函數輸入輸出的特性統一且分開,相對於原生的函數,常常因為輸入的資料格式不同,也會輸出不同的結果,這某些方面的“聰明”,在purrr中都簡潔化,但相對的就會產生很多相同功能,但對應不同資料格式的函數如map_chr(), map_dbl()等。
2. 將pipe %>% 的適用性導入purrr中
3. 改善function error handling的機制(使用safely())
這邊收集相關的發佈資料,能更理解這purrr項目相關進展的脈絡!(把purrr package稱為項目,是覺得這package的開發很有野心,想要把R語言在往可以適應於分散式運算系統,且整個代碼也是在一整個tidyverse下的邏輯開發的,很有一貫性)
purrr 0.1.0 2015/9/29
purrr 0.2.0 2016/1/06








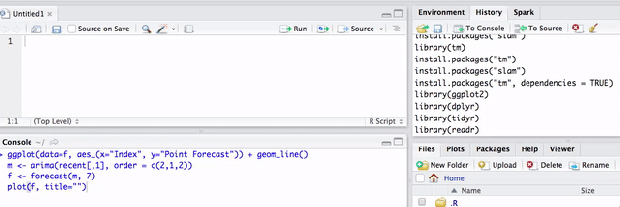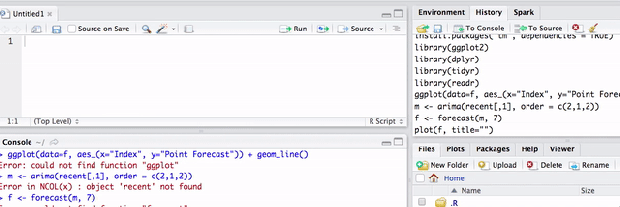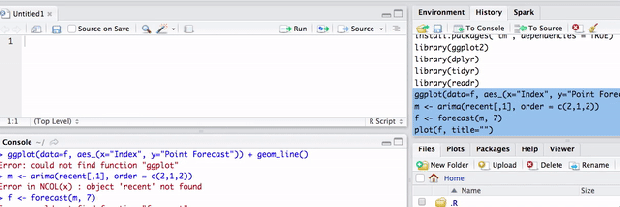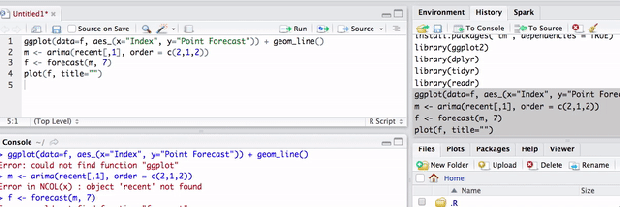
 的
的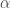 來判斷,也就是控制所謂的Type I Error(不應該reject NULL hypothesis但卻reject了),而我們常常取所謂的p value < 0.05 % 來代表統計上有顯著意義,其實是說因為“搞錯的機率小於5%,所以應該沒問題的概念,簡化為猜一百次,可能有五次猜錯”,而當我們在同一個樣本中,同時比了上萬次,那就代表我們必定會挑出很多p value有顯著,但其實是“搞錯了”的基因,此類問題其實就是所謂的Multiple Comparison 問題
來判斷,也就是控制所謂的Type I Error(不應該reject NULL hypothesis但卻reject了),而我們常常取所謂的p value < 0.05 % 來代表統計上有顯著意義,其實是說因為“搞錯的機率小於5%,所以應該沒問題的概念,簡化為猜一百次,可能有五次猜錯”,而當我們在同一個樣本中,同時比了上萬次,那就代表我們必定會挑出很多p value有顯著,但其實是“搞錯了”的基因,此類問題其實就是所謂的Multiple Comparison 問題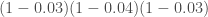

 的展開:
的展開: