最近稍微熟悉系統分析的方法後,希望把這套方法學往臨床上去找主題做應用,無意間發現內科學聖經哈里遜內科學最新版新增了這個章節:Network Medicine: Systems Biology in Health and Disease,是由哈佛醫學院Joseph Loscalozo心臟內科醫師所撰寫,非常的令人驚喜!出自臨床醫師之手,且寫得淺顯易懂,點出了其中一些方向,用pathobiology的觀點來應用系統生物學的方法分類臨床疾病(雖然裡面所引用的論文大概是5-10年前的文獻)。
這篇87e章節的重點可分成三大部分:
1. 生物的複雜系統(complex biology system)觀念介紹
2. 病態生理學和系統生物學共通的精神
3. 系統醫學(network medicine)重新定義疾病的看法
基本上,對於一個疾病的掌握,本來就是系統性著手的(對於一個疾病的診斷、監測、治療,在臨床上都是多角度切入的),如何導入更“quantitative”的方法和工具,則是其中能進步的關鍵點,而非為了“系統生物學”而”系統生物學“。
System biology, which is defined as the holistic study of living organisms or their cellular or molecular network components to predict their response to pertubations
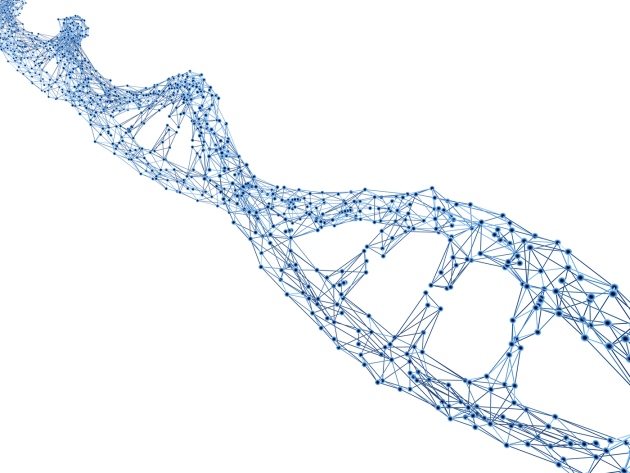
分析一個複雜的生物系統(complex biology system)被擾亂(pertubation)或是影響後的變化,可以藉由觀察這個系統的架構(topology)和其可能反應來著手,這個系統可以看成是一個由不同大小模組(modules)所組成的網絡(network),其中簡化成所謂的節點(nodes)和連結(links),整體系統組成的功能並非單純局部模組功能的疊加。
不同連結方式所產上的網絡,特性也不同,而生物系統的連結方式是所謂的"scale-free"的,簡單說,就是只有少數的結點連結數很高,大多數節點的連結數偏低,從數學上來描述的話,其節點和連結束的對數是呈線性的(power law),可以參考下面的圖

左邊的網絡是每個節點隨機的跟另一個節點相連結,結果不同聯結數量的節點呈現鐘型分佈(poisson distribution),右邊則是我們所說的生物網絡連結方式,少數節點擁有高聯結數,多數節點連結數很低。
scale-free network的特性有什麼呢?
第一點,其有許多冗余功能的節點於網絡中(redundancy),無形中讓系統非常穩定,除非這些高度連結的節點受到影響。
第二點,這樣網絡的冗余特性提供了生物演化上的特質,能容忍許多不同的基因變異或是調控上的微小差異
第三點,scale-free的網絡,任意兩點間的連結路徑其實是短的,只要經由約莫6-7個節點便能連結兩個節點,這樣的性性相對於所謂的隨機形成網絡,其在傳遞特殊訊息時候是非常有效率的。
系統生物學應用於病態生理學
近十年開始陸續有許多系統生物學的方法被使用在一些特定疾病的病生理過程上,這篇文章有提到下面四種疾病糖尿病、遺傳性運動失調、EBV感染、肺動脈高壓,其分別使用了系統生物學的概念去重新理解疾病的機制。
遺傳性運動失調
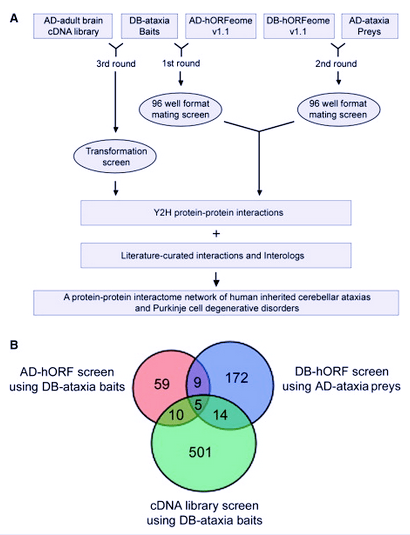
在這篇2006年的研究,主要探討 cerebellar Purkinje cell (PC) degeneration,先做基本文獻回顧,找出目前已知跟這疾病相關的53個可能致病蛋白質(來自23種遺傳性運動失調疾病),再使用yeast-two hybrid的方式來做蛋白質-蛋白質互動(protein-protein interaction)的研究,並且發這些可能致病蛋白質間的關聯,已便更理解這複雜疾病的可能致病機轉。
糖尿病
這篇2012年的研究,主要探討糖尿病前期(pre-diabetes)的相關生物標識,使用KORA這個德國大型研究計劃中的個案檢體(KORA,Cooperative Health Research in the Region Augsburg, 在德國奧斯陸地區的族群健康研究,從1996年開始,大約持續10年),同時將這些檢體拿去做基因表現量和代謝體的檢測,代謝體檢測是使用在整合這兩種資料來探討可能用來預測跟糖尿病前期相關的生物標識
EBV/HPV病毒感染
這篇2012年的研究,使用了非常豐富的研究方法,想探討EBV/HPV感染所造成的宿主反應,跟相關疾病的關聯,使用了EBV-human Y2H 和HPV-human Y2H library來做相關的protein-protein互動的資料,接者收集EBV-human/HPV-human組織的基因表現資料,最後還收集了美國相關感染過EBV/HPV病毒的患者資料,來看其造成其他相關疾病的風險變化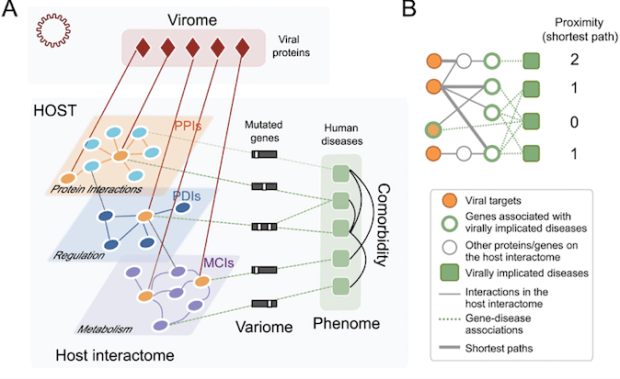
肺部動脈高壓
這篇也是2012年發表的研究,探討microRNA-21對於肺部高壓的調控關係,這篇一開始先用文獻檢索,找到相關可能的已知跟肺高壓有關的基因,再針對這些基因的蛋白產物去做protein-protein interactome的分析,使用“consolidated database”(看完這篇文章的supplement,其實裡面對於分析細節揭露的很不清楚,稍微失望)。
總結:
在內科聖經哈里遜上能看到network medicine的章節,的確代表這類型的新分析方式將慢慢能用來解決臨床問題,另一方面,這篇文章所引用的文章大概是5-10年前的研究,這幾年許多新的資料庫出現,從生物路徑到蛋白質-蛋白質互動資料庫,且分析方法越來越成熟,可見這類分析方式會更快地被帶入到臨床研究之中。
參考閱讀:
Lim et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125:801-814, 2006
Wang-Sattler et al. Novel biomarkers for pre-diabetes identified by metabolomics.Mol Syst Biol 8:615, 2012
Gulbahce et al. Viral perturbations of host networks reflect disease etiology.
PLoS One 8:e1002531, 2012
Victoria N. Parikh et al. MicroRNA-21 Integrates Pathogenic Signaling to Control Pulmonary Hypertension: Results of a Network Bioinformatics Approach. Circulation. 2012 Mar 27; 125(12): 1520–1532.
Parikh et al. Towards Systems Biology of Pulmonary Hypertension. Circulation 125:1520-1532, 2012
KI Goh et al. The human disease network. Proc Natl Acad Sci USA 104:8685, 2007
J Loscalzo et al. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Molec Syst Biol 3:124, 2007˙