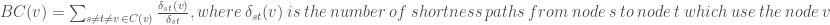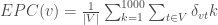主要是想寫一個程式來當作媒介,來做一些並行運算的設計(當然,假如已經知道整體計算細節和運算的資料,也許可以考慮使用hadoop或是spark這些專門針對分散式資料的數據運算設計的框架),這邊算是土炮來練習如何用python來加速數據運算的框架,藉此熟悉python底層在thread和process的設計,最後這邊使用多進程(process)設計,搭配sqlite3在中間儲存資料,過程呼叫netMHC來進行單組fasta資料的運算。
實作過程發現的一些小心得和重點,把掉坑心得分享一下:
1. 理解sqlite3資料庫的特性
sqlite資料庫的輕特性很適合用來做數據處理原型的資料儲存用,但他在transaction rate限制下要在代碼中考慮進去,否則會成為程式速度很大的瓶頸,雖然sqlite資料庫其實是能支援多thread來讀取的,可是要在compile sqlite安裝時去修改原始代碼,對於不太熟悉其底層的人來說會很困難。
2. python語言的特性
python底層設計上有一個global interpreter lock的設計,所以當你所有程式都是python代碼的時候,即使使用python內的multiprocess或是multithreading,效果依舊有限,可是把python當作是控制主程式,用多線程或是進程來控制一些主要運算,這些運算是用其他語言小程式執行(shellscript, java 等外部程式),效果就會不錯。
3. 理解python3在tmp檔上之實作細節
在調用python3的tempfile模組時,要記得背後的原理,否則會發現當你開啟幾千個線程去使用temp黨時,名字強蹦的機會還是有的,這時候會頗嚴重的。
4. 理解processing和threading的差異及處理
processing和threading在python中的使用方式和語法其實非常相似,主要是底層資源的隔離或是溝通的差異,這兩種不一定是哪一種就比較好,要看自己計算上的瓶頸是什麼,是網路等待貧頸,或是資源調用大量等,頗多底層細節和對自己計算過程的掌握相關,其實真的需要經驗,也是這次採坑才知道,也才理解搭建出hadoop/spark框架的人真的頗有價值。
5. 理解在使用Multiprocessing.queue和Queue.queue的差異
在測試過程會發現總會有幾個進程會無緣無故地停著不動,但也沒有進入下一個任務,後來才發現queue模組中的細節,比如像下面這個語句
def doJob(*args):
que = args[0]
conn = args[1]
process_name = multiprocessing.current_process().name
while que.qsize() > 0:
try:
job = que.get(timeout=5)
job_num = que.qsize()
job.do(conn, process_name, job_num)
except:
print("".join(["Queue is empty"]))
print("".join(["****",process_name,"do a good job, let it take a break","***"]))
在上面的語法中,用que.qsize來作為thread/process結束時候的信號,讓他不會進行去跟queue要物件來執行,但後來發現在多進程或多線程下,有可能在做while中判斷時qsize是大於0,下一剎那當queue內物件已經被其他線程或是進程取完後,當他進入到queue.get時,預設是會一直等到有物件,除非設置timeout的參數,設置之後就可以讓這些不小心掉如無主狀態的進程或是線程歸位了!
import subprocess
import tempfile
import re
import sqlite3
import os
import time
import queue
import threading
import multiprocessing
def createdatabase(choose_HLA_type_super):
"""
create the database and table with the HLA type
"""
database_name = "{database}_netMHC.db".format(database=re.sub('-','_',choose_HLA_type_super))
# create the table in the database
conn = sqlite3.connect(database_name)
c = conn.cursor()
# Create different table according to peptides length
# Create table
c.execute('''CREATE TABLE Neoantigen_14
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_13
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_12
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_11
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_10
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_9
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
c.execute('''CREATE TABLE Neoantigen_8
(pos integer, HLA text, peptide text, Core text, Offset integer, I_pos integer, I_len integer,
D_pose integer, D_len integer, iCore text, Identity text, llog50k real, Affinity real, Rank REAL, BindLevel integer)
''')
# Save (commit) the changes
conn.commit()
# We can also close the connection if we are done with it.
conn.close()
#print(''.join(['Database ', database_name,' created!']))
# string 14 - 9
def peptideshorten(peptide):
"""Prepare the peptides for 14 - 8 lengths affinity predict
Args:
seq
Returns:
A Generator gave the trim peptides from 14 - 8
Raises:
"""
i = 0
yield peptide
while i < 6: i += 1 yield peptide[i:-i] def stringtrim(peptide): """Trim the string to remove spaces Args: seq Returns: A string without spaces Raises: """ return re.sub('\s','',peptide) # Input with single peptide per time def netMHCcalculate(single_peptide_name, single_comment, single_peptide, predict_list, choose_HLA_type_super): #print("Begin to predict with netMHC!") while True: try: single_peptide_window = next(single_peptide) peptide_seq_len = len(single_peptide_window) peptide_len = str(int((peptide_seq_len + 1)/2)) one_record = ''.join(['>',single_peptide_name,' ',single_comment,'\n',single_peptide_window])
#print(''.join(['The whole peptides length: ', str(peptide_seq_len)]))
#print(''.join(['The Peptide Length for prediction: ', peptide_len]))
#print(one_record)
f = tempfile.NamedTemporaryFile(mode='w+',delete=True, suffix='.fasta')
f.write(one_record)
f.seek(1)
predict_list.append(subprocess.check_output(['sh', 'netMHC_one.sh', choose_HLA_type_super, peptide_len, f.name]))
f.close()
except StopIteration:
break
def multplerecords_netMHCcalculate(multiple_records_iter, choose_HLA_type_super):
i = 0
predict_list = []
while True:
try:
single_record_fasta = ''.join(next(multiple_records_iter))
f = tempfile.NamedTemporaryFile(mode='w+',delete=True, suffix='.fasta', prefix=''.join(['tmp',str(round(time.time()))]))
f.write(single_record_fasta)
f.seek(1)
predict_list.append(subprocess.check_output(['sh', 'netMHC_one.sh', choose_HLA_type_super, str(14-i), f.name]))
f.close()
i += 1
except StopIteration:
break
return(predict_list)
def get_single_row_predict(predict_iter):
single_row_predict = []
while True:
try:
single_result_list = iter(next(predict_iter).decode().split('\n'))
while True:
try:
single_result = next(single_result_list)
if len(single_result) == 0:
continue
single_row_predict.append(single_result)
except StopIteration:
break
except StopIteration:
break
return(single_row_predict)
# Seperate the conn, reduce the frequence to open a new connection
def update_insert_database(single_row_predict_iter, conn):
print("Begin to insert result into database")
c = conn.cursor()
one_larger_trasaction_14 = []
one_larger_trasaction_13 = []
one_larger_trasaction_12 = []
one_larger_trasaction_11 = []
one_larger_trasaction_10 = []
one_larger_trasaction_9 = []
one_larger_trasaction_8 = []
while True:
try:
single_one_row_predict = next(single_row_predict_iter).split(',')
#print(single_one_row_predict)
D_POS, D_HLA, D_Peptide, D_Core, D_Offset, D_I_pos, D_I_len, D_D_pos, D_D_len, D_iCore, D_Identity, D_log50k, D_Affinity, D_Rank, D_BindLevel = single_one_row_predict
if len(stringtrim(D_Peptide)) == 14:
one_larger_trasaction_14.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 13:
one_larger_trasaction_13.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 12:
one_larger_trasaction_12.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 11:
one_larger_trasaction_11.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 10:
one_larger_trasaction_10.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 9:
one_larger_trasaction_9.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
if len(stringtrim(D_Peptide)) == 8:
one_larger_trasaction_8.append((int(D_POS), re.sub('\s','',D_HLA), re.sub('\s','',D_Peptide), re.sub('\s','',D_Core), int(D_Offset), int(D_I_pos),int(D_I_len), int(D_D_pos), int(D_D_len), re.sub('\s','',D_iCore), re.sub('\s','',D_Identity), float(D_log50k), float(D_Affinity), float(D_Rank), float(D_BindLevel)))
except StopIteration:
# close the database
#print(''.join(['Insert data into database, finished!']))
break
c.executemany("INSERT INTO Neoantigen_14 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_14)
conn.commit()
c.executemany("INSERT INTO Neoantigen_13 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_13)
conn.commit()
c.executemany("INSERT INTO Neoantigen_12 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_12)
conn.commit()
c.executemany("INSERT INTO Neoantigen_11 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_11)
conn.commit()
c.executemany("INSERT INTO Neoantigen_10 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_10)
conn.commit()
c.executemany("INSERT INTO Neoantigen_9 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_9)
conn.commit()
c.executemany("INSERT INTO Neoantigen_8 VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)", one_larger_trasaction_8)
conn.commit()
#conn.close()
class Job:
def __init__(self, sub_name_list, sub_comment_list, sub_seq_list, multiple_fasta_num, HLA_type):
self.sub_seq_list = sub_seq_list
self.sub_name_list = sub_name_list
self.sub_comment_list = sub_comment_list
self.HLA_type = HLA_type
self.multiple_fasta_num = multiple_fasta_num
def do(self, conn, process_name, job_num):
print(''.join([process_name,": Begin ",str(job_num),"_th Job!"]))
multiple_records = create_multiple_records(self.sub_name_list, self.sub_comment_list, self.sub_seq_list, self.multiple_fasta_num)
print(''.join(['_',process_name,"_Finish create multiple recors!"]))
multiple_records_iter = iter(multiple_records)
print(''.join(['_',process_name,"_create multiple record iterator"]))
# calculate the netMHC on single peptite with 8 - 14 peptide and single HLA type
predict_list = multplerecords_netMHCcalculate(multiple_records_iter, self.HLA_type)
print(''.join(['_',process_name,"_Finish netMHCcalculate"]))
predict_iter = iter(predict_list)
print(''.join(['_',process_name,"_Create precit_list iterator"]))
# turn calculate result into single string for per sequence affinity prediction
single_row_predict = get_single_row_predict(predict_iter)
print(''.join(['_',process_name,"_Create single row preidcit data"]))
single_row_predict_iter = iter(single_row_predict)
# insert the data into database
print(''.join(['_',process_name,"Prepare to insert database"]))
update_insert_database(single_row_predict_iter, conn)
print(''.join(["_",process_name," Job", "calculate finished!"]))
def doJob(*args):
que = args[0]
conn = args[1]
process_name = multiprocessing.current_process().name
while que.qsize() > 0:
try:
job = que.get(timeout=5)
job_num = que.qsize()
job.do(conn, process_name, job_num)
except:
print("".join(["Queue is empty"]))
print("".join(["****",process_name,"do a good job, let it take a break","***"]))
def seperateIterate(iter_item, multiple_fasta_num):
i = 0
sub_item_list = []
while i < multiple_fasta_num:
sub_item_list.append(next(iter_item))
i += 1
return sub_item_list
HLA_type_super=["HLA-A0101","HLA-A0201","HLA-A0301","HLA-A2402","HLA-A2601","HLA-B0702","HLA-B0801","HLA-B2705","HLA-B3901","HLA-B4001","HLA-B5801","HLA-B1501"]
HLA_type_all_ABC=["HLA-A0101","HLA-A0201","HLA-A0202","HLA-A0203","HLA-A0205","HLA-A0206","HLA-A0207","HLA-A0211","HLA-A0212","HLA-A0216","HLA-A0217","HLA-A0219","HLA-A0250","HLA-A0301","HLA-A1101","HLA-A2301","HLA-A2402","HLA-A2403","HLA-A2501","HLA-A2601","HLA-A2602","HLA-A2603","HLA-A2902","HLA-A3001","HLA-A3002","HLA-A3101","HLA-A3201","HLA-A3207","HLA-A3215","HLA-A3301","HLA-A6601","HLA-A6801","HLA-A6802","HLA-A6823","HLA-A6901","HLA-A8001","HLA-B4403","HLA-B4501","HLA-B4601","HLA-B4801","HLA-B5101","HLA-B5301","HLA-B5401","HLA-B5701","HLA-B5801","HLA-B8301","HLA-B0702","HLA-B0801","HLA-B0802","HLA-B0803","HLA-B1402","HLA-B1501","HLA-B1502","HLA-B1503","HLA-B1509","HLA-B1517","HLA-B1801","HLA-B2705","HLA-B2720","HLA-B3501","HLA-B3503","HLA-B3801","HLA-B3901","HLA-B4001","HLA-B4002","HLA-B4013","HLA-B4201","HLA-B4402","HLA-B5802","HLA-B7301","HLA-C0303","HLA-C0401","HLA-C0501","HLA-C0602","HLA-C0701","HLA-C0702","HLA-C0802","HLA-C1203","HLA-C1402","HLA-C1502","HLA-E0101"]
result_title = ['pos', 'HLA', 'peptide', 'core', 'offset','I_pos','I_len', 'D_pos','D_len', 'iCore', 'Identity', 'log50k', 'Affinity','Rank','BindLevel']
# Parse the fasta into program
tabule_fasta = subprocess.check_output(['sh', 'TurnTabule.sh', 'cosmic_neoantigen_tmp.fasta'])
tabule_fasta_list = tabule_fasta.decode().split('\n')
# Splite the singel record into name, comment, seq
name_list = []
comment_list = []
seq_list = []
for fasta in tabule_fasta_list:
try:
name, comment, seq = fasta.split('\t')
name_list.append(name)
comment_list.append(comment)
seq_list.append(seq)
except ValueError:
print(fasta.split('\t'))
choose_HLA_type_super = HLA_type_super[0]
# create multiple database for each threads
database_create_list = []
process_num = 100
for i in range(process_num):
database_create_list.append(''.join([choose_HLA_type_super,"_",str(i)]))
# create the database
for database_name in database_create_list:
createdatabase(database_name)
database_name_list = []
for i in range(process_num):
database_name_list.append(''.join([re.sub('-','_',choose_HLA_type_super),'_',str(i),"_netMHC.db"]))
database_conn_list = []
for i in range(process_num):
conn = sqlite3.connect(database_name_list[i])
database_conn_list.append(conn)
# use iterator to decrease memory use
iter_name = iter(name_list)
iter_comment = iter(comment_list)
iter_seq = iter(seq_list)
# que_list
# change to use the multiprocessing.queue
# emerge the que to one
que_list = []
work_num = 3573
multiple_fasta_num = 1000
i = 0
que = multiprocessing.Queue()
while i < work_num:
try:
sub_name_list = seperateIterate(iter_name, multiple_fasta_num)
sub_comment_list = seperateIterate(iter_comment, multiple_fasta_num)
sub_seq_list = seperateIterate(iter_seq, multiple_fasta_num)
que.put(Job(sub_name_list, sub_comment_list, sub_seq_list, multiple_fasta_num, choose_HLA_type_super))
i += 1
except StopIteration:
break
start_time = time.time()
process_list = []
for i in range(process_num):
p = multiprocessing.Process(target=doJob, name=''.join(['Process_',str(i)]), args=(que, database_conn_list[i]))
process_list.append(p)
print(p)
p.start()
for p in process_list:
p.join()
print(''.join(["done!, take",str(time.time()-start_time), " sec"]))





![MNC(v)=|V(MC(v)|, where MC(v) \ is \ a \ maximum \ connected \ component\ of\ the\ G[N(v)]](https://s0.wp.com/latex.php?latex=MNC%28v%29%3D%7CV%28MC%28v%29%7C%2C+where+MC%28v%29+%5C+is+%5C+a+%5C+maximum+%5C+connected+%5C+component%5C+of%5C+the%5C+G%5BN%28v%29%5D+&bg=ffffff&fg=404040%3B&s=0&c=20201002)