最近對於所謂的“Annotation”這個辭彙在生物資訊裡的意思越來越有感覺,而且深深覺得其對於整個生資分析中所佔有極重要的角色,經驗高低可能就決定在此,很多基本的分析流程都是可以很快速學會的,但是對於自己資料做好的annotation卻是沒有固定模式可學的。
那什麼是annotation的定義呢?(這邊是在探討比較廣義層面的)
下面是annotation在wiki裡面的定義,簡單而精準:
An annotation is a metadata attached to text, image, or other data. Often, annotations make reference to a specific part of the original data.
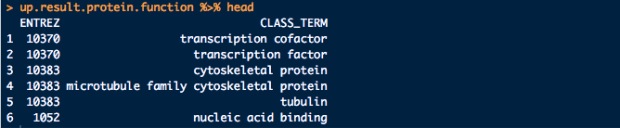
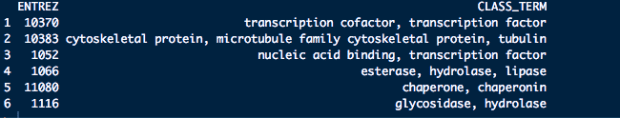
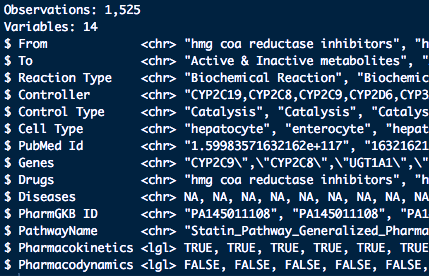
好的annotation其實就是善用全世界科學家花費時間和金錢做實驗得出的寶貴資料,將其應用在自己的分析專案中,怎麼善用全世界上千個資料庫的資訊,就必須要有廣泛的知識,且另一方面,進行annotation的過程,本身就有一些“目的”導向,通常你就要很清楚自己想要了解的方向,比如你拿到了一個Gene list,你好奇的是這群基因的序列關係、功能上的關係、藥物反應、多少比例是文獻中跟癌症幹細胞相關的標記、多少是跟生長因素相關的、分別牽涉到什麼生物路徑的,根據不同的好奇方向,其實就要去找不同的資料庫來佐證,且怎麼呈現又是另一門需要花費心思的事情。
從bioconductor上面的package總數,有一半以上都是跟annotation功能相關的

Bioconductor的計劃其中很重要的便是讓R變成在做生物資訊annotation的專業工具,所以開發出很多的annotation package來達成這個目的,且有一套良好的規範,分別讓想要將資料庫包成package,或是如何好的query資料,都能更加的統一性,其中AnnotationDbi就是其中一個package,將各種可以用來註釋的資料包成package,不用再使用web request的方式,這結構便是由AnnotationDbi packages所決定的,而這其中分成很多種類:
- Organism level: e.g org.Mm.eg.db
- Platform level: e.g. hgu133plus2.db
- Homology level: e.g. hom.Dmi.inp.db
- Transcriptom level:e.g.TxDb.Hsapiens.UCSC.hg19.knownGene,EnsDb.Hsapiens.v75
- System-biology level: GO.db

這結構很好的把所有在bioconductor中的annotation資源分成五大類,並能根據這五大類來找尋自己所需要用的annotation 資源,而不會被搞糊塗。