Tophat介紹與語法
介紹
Tophat是一款可以用來拼基因序列的生資工具,其最重要的特色就是其演算法對於mRNA的splicing junction有比較好的辨識能力。
由約翰霍普金斯大學的Daehwan Kim 和 Steven Salzberg ( Center for Computational Biology)以及 華盛頓大學的Cole Trapnell 教授( Genome Sciences Department ). 在約翰霍普金斯大學有這款工具的詳細指南, 這工具主要是在linux下的command line執行。
原理
這邊介紹此工具在比對時的原理,可以幫助思考在設計流程的細節,以下是圖解:

基本運作原理:
- 第一步:Optimal transcriptome alignment
- 第二步Genome alignment
- 第三步:Spliced alignment
在Paired-end的RNAseq裡面:
- 假如有Annotation的話,其會先利用bowtie2 genome index 由gtf/gff file組成transcript sequences,先將Reads針對其來ALIGNMENTS
- 當reads對不到Transcripts時,其會跟genome mapping
- 此時會將ummaped reads分成小片段在去對genome mapping,此時其會根據我們設定的最大intron size來跑分析,此時tophat會同時尋找indel或是fusion break point
- 基因序列在可能splicing site的地方會被index且concatenate,此時沒有mapped到哦會被放在junction flanking index
- Segment alignments 被stitch起來行成完整的alignment
- 當multiple alignment發生時,tophat會計算其分別的alignment score
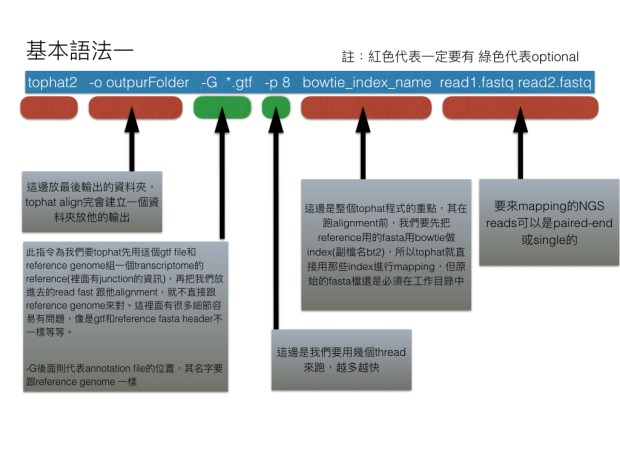
語法