在處理基因表現差異的時候,通常都是要比較兩個生物樣品間,幾萬個基因有哪些是有表現差異的,此時就會同時做多次檢定,此時就會遇到一個很有趣的問題!
因為當我們在針對單一個基因在實驗設計下,是否有表現差異時,通常使用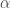
那我們如何評估經過多重比較後,我們必定會搞錯一次的機率呢?
此時就可以使用Family-wise error rate,概念上其實很簡單,把每個檢體中基因間顯不顯著,當作是獨立事件,而其p值,就代表者“搞錯的機率”,假設我們有三個基因A,B,C,同時做檢定,分別得到0.03, 0.04, 0.03,以A基因為例(假設null hypothesis為兩個基因沒有表現差異),當我們要推翻這個假設時,可能有0.03的機率可能無法推翻,換句話說,我們有1-0.03的機率篤定他是沒問題,可以大方推翻檢定的,所以當我們把每個基因間當作獨立事件來看,我們一定沒錯的機率大概是:
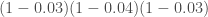
反之,我們一定至少有一個基因,其實沒有顯著差異的機率(在這也就是Family-wise error rate):
$latex FWER = 1 – (1-0.03)(1-0.04)(1-0.03) $
所以當我們可以評估多重比較後,出錯的機率後,下一步當來就是來調整原本單個基因是否表現差異,只用p = 0.05就顯著的策略,改成隨者我們同時比較的基因數量越多,我們要越嚴格一些,根據 FWER的概念,所衍生出來的調整策略,主要有下面三種:
1.Sid´ak procedure
此方法是由比較直覺的方式來處理多重檢定的問題
$latex\alpha_{c} = 1 – \sqrt[R]{1-\alpha_{e}} $
$latex\alpha_{c},the probability of a Type I error at the gene level$
$latex\alpha_{e},the probability of a Type I error at the experiment error$

2.Bonferroni
Bonferron的方式主要是建立在Sidak correction上,只是其簡化原本sidak correction中的
因此可以簡化為下面的算式
3.Holm’s method(Stepwise Algorithm)
但可以發現,這種調整方式,有點暴力,大量基因多重檢定的時候,要顯著的p值會順間變得很嚴格。
相對於FWER,因為其是假設所有基因間是獨立關係而來的數值,但實際上生物體內基因間,多少都會有相關連,所以目前另一種用來作為多重比較的指標是False Discovery Rate(FDR)

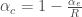
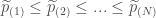

跟其衍生而來的方法則有:
1.Benjamini & Hochberg
2.Benjamini & Yekutieli
在R裡面可以簡單使用
p.adjust(p, method = p.adjust.methods, n = length(p))
p.adjust.methods
# c("holm", "hochberg", "hommel", "bonferroni", "BH", "BY",
# "fdr", "none")
在處理像是pathway analysis或是microarray analysis時,使用Bonferroni, Sidak and Holm’s corrections顯得太過保守且沒考慮到生物上的意涵。目前認為使用FDR(False Discovery Rate)的方式相對恰當,以此觀念出發其實也發展出很多FDR的變化,每個都有些微不同的假設,包括Benjamini-Hochberg correction、Benjamini-Yekutieli correction, positive FDR、adaptive Benjamini-Hochberg correction、significant analysis of microarrays, Storey’s q-value, resampling based approach.這之中以Benjamini-Yekutieli方法最為保守。
關於這方面的知識,推薦這本書Bradley Efron, Large-Scale Inference: Empirical Bayes Methods for Estimation, Testing and Predition, Stanford University

Good introduction! Thanks a lot. I’m working on a relevant note: https://hackmd.io/3ixhWBkxQZe2op-VtGq42g?view
讚讚