topGO package在bioconductor裡已經超過10年了,算是最早R裡面可以用來做GO term enrichment analysis的工具,但也因此裡面處理輸入資料的方式相對的來說頗不方便,語法比較接近R base,所以對於用習慣tidyverse體系的package的人來說會有點頭痛,這邊則來分享一下,如何使用topGO來做GO analysis
使用topGO做GO analysis主要有五個步驟:
1. 將基因列表轉換為Entrez ID
2. 使用org.Hs.eg.db資料庫分別找出每個Enrez ID所對應到的GO terms
3. 將資料結構處理成topGO中所使用的topGOdata object
4. 使用runTest進行GO analysis
5. 使用GenTable來獲取結果成data.frame
最麻煩的地方是理解topGOdata object建立所需要的常數,和每個輸入的結構!
這邊補充一下,各個下面代碼的重要資料結構長得樣子
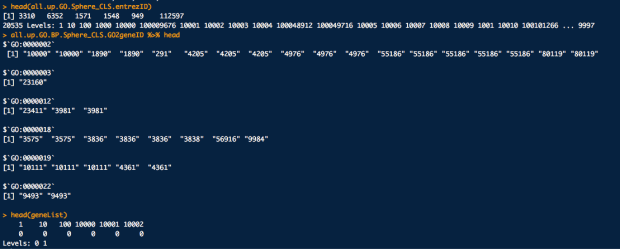
library(annotationDbi)
library(org.Hs.eg.db)
library(purrr)
library(dplyr)
library(topGO)
all.up.GO.Sphere_CLS.entrezID <- up.tukey.aov.Sphere_CLS %>% with(EntrezID)
#使用annotationDbi中提供的select方法,query的結果為data.frame的形式
all.up.GO.Sphere_CLS <- AnnotationDbi::select(org.Hs.eg.db, keys = all.up.GO.Sphere_CLS.entrezID [!is.na(all.up.GO.Sphere_CLS.entrezID )] %>% as.character,
columns = "GO",
keytype = "ENTREZID")
#將entrezID對應GO term的data.frame格式轉換成list-like結構
all.up.GO.Sphere_CLS.list <- all.up.GO.Sphere_CLS %>%
filter(EVIDENCE != "IEA") %>% split(.$ONTOLOGY, .$ENTREZID)
all.up.GO.BP.Sphere_CLS <- all.up.GO.Sphere_CLS.list$BP
all.up.GO.BP.Sphere_CLS <- all.up.GO.BP.Sphere_CLS %>% split(.$ENTREZID) %>% map(with, GO)
all.up.GO.BP.Sphere_CLS.GO2geneID <- inverseList(all.up.GO.BP.Sphere_CLS)
#建立topGO所需要輸入參數allGenes, gene2GO兩個輸入變數geneList,all.up.GO.BP.Sphere_CLS
geneNames <- names(all.up.GO.BP.Sphere_CLS)
geneList <- factor(as.integer(geneNames %in% (up.Sphere_CLS$EntrezID[!is.na(up.Sphere_CLS$EntrezID)] %>% as.character)))
names(geneList) <- geneNames
# construct topGOdata
GOdata <- new("topGOdata", ontology = "BP", allGenes = geneList,
annot = annFUN.gene2GO, gene2GO = all.up.GO.BP.Sphere_CLS)
# calculate the result
resultFisher <- runTest(GOdata , algorithm = "classic", statistic = "fisher")
resultKS <- runTest(GOdata, algorithm = "classic", statistic = "ks")
resultKS.elim <- runTest(GOdata, algorithm = "elim", statistic = "ks")
# query the result
allRes <- GenTable(GOdata, classicFisher= resultFisher,
classicKS = resultKS, elimKS = resultKS.elim,
orderBy = "classicFisher", ranksOf = "classicFisher", topNodes = 200)
allRes <- allRes %>% transmute(GO.ID=GO.ID,
Term=Term,
Annotated=Annotated,
Significant=Significant,
Expected=Expected,
classicFisher=as.numeric(classicFisher),
classicKS=as.numeric(classicKS),
elimKS=as.numeric(elimKS))
allRes.up.Sphere_CLS <- allRes %>% filter(classicFisher < 0.05)
