
Gene Set Enrichment Analysis的方法是目前在pathway analysis方法中,歸類為functional score analysis裡的state of the art(可參考此篇),且其搭配的工具和gene sets都有蠻完整的支持,值得深入瞭解,並納為自己的口袋武器。實際上,Gene Set Enrichment Analysis有許多係分支的方法,但這邊主要談的是由Broad Institute裡的GSEA這款軟體。
基本觀念
所有的pathway analysis的觀念都是用一組特定的gene set簡化代表特定生物內的pathway或是功能,然後看這組gene set是否有在我們的資料中有特別顯著的表現。
不管是在做pathway analysis或是gene set enrichment analysis(其實大同小異,做時候的觀念差異不同而已),到後來會發現如何挑選gene set也變成是一種功夫,必須依靠對背景的生物問題有透徹的文獻研究才會能有較詳盡的分析結果。可以研讀Hammer lab中對於自制gene list的一些看法,會對觀念的釐清有頗大的幫助,其實在做pathway analysis的本身就是一種生物高通量資料探勘的過程。
要如何做GSEA呢?
(這邊以由Broad institue維護的GSEA使用來分享)
第一步理解GSEA所需要輸入的資料格式:*.gct, *.cls
GCT:Gene Cluster Text
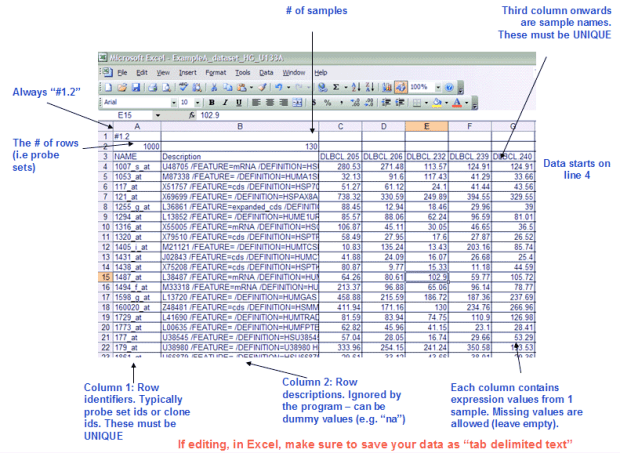
CLS: Categorical class

第二步挑選或是設定一組基因列表S(關鍵在於priori defined)
這群基因可以代表某個metabolic pathway或是共有特定的GO term,想要測定這個特定的pathway或是go function group有沒有在整個L基因列表中有表現顯著,在GSEA論文中所使用的gene set內大小至少35個gene,不過很多人自製的gene set並不一定會超過這數量。(在GSEA的軟體裡能直接抓取MSigDB資料庫 )
)
第三步製作一個根據基因在兩組phenotype中表現值差異來排序的基因列表
量測兩組樣本間某個基因表現量的差異,有許多作法,在GSEA官方軟體內所使用的主要有五個指標,用來表現基因在兩個表型間的差異,因為之後排序就是依照這些值,所以最好理解一下他們各自的算法,分別是:Signal2Noise(預設方法), tTest, Ratio_of_Classes, Diff_of_Classes, log2_Ratio_of_Classes。
下面則是他們的公式:
Signal2Noise
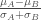
tTest
這方法至少每一組的樣本都要有三個會比較適合

Ratio_of_Class
其實就是所謂的Fold Change,兩者差異越大,數值就會偏離1

Diff_of_Classes
直接用兩者的差值
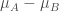
log2_Ratio_of_Classes
將計算好的fold change取對
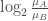
第四步計算Enrichment Score(ES)
整個GSEA分析中,最關鍵的觀念就是Enrichment Score的算法理解,在排序好的基因列表L中,我們所定義的S genes列表中的基因理論上會散佈在其中,此時由左到右,計算累計的統計量(running-sum statistic),假如遇到的基因是在S genes中的,那麼就加


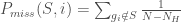
第五步計算ES有無顯著
會發現這其實就是一種修改過的無母數統計做法,比較重要的是他比較的是當我們隨機把phenotype的標籤做置換,然後經過permute後,比較兩者有無差異的方式。
第六步多重比較的事後調整(adjustment)
當我們有很多組S Genes要看有無顯著時,這時候就有多重比較的問題,這邊主要使用False Discovery Rate來控制。
其實假如直接下載使用Broad Institute維護的軟體GSEA話,只要做第一個步驟就行了,輸入資料後接下來就不用擔心了,這是理解這六步驟才能真的懂GSEA分析的邏輯。
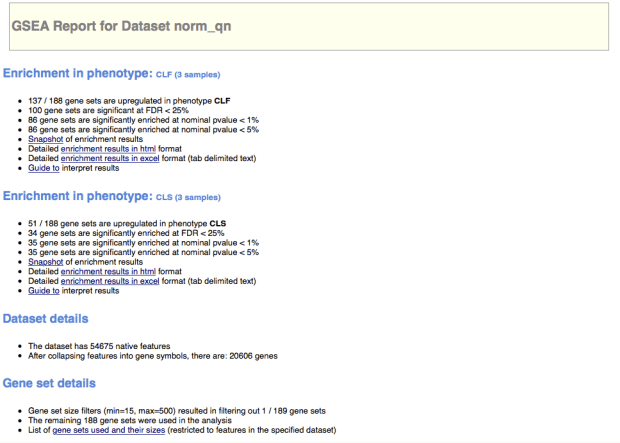
下面展示一些使用GSEA分析後的輸出檔案,其實幫助很大,裡面內建的gene set非常多,也可以由這些gene set來建立一些生物感覺。

 閱讀參考:
閱讀參考:
Aravind Subramanian, Pablo Tamayo, Vamsi K. Mootha et al.(2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS, vol 12, 15545-15550
