做複雜的次世代定序資料分析,少不了使用基因組瀏覽器來(Genome Browser),基因組瀏覽器是圖形化的工具用來展示生物序列資訊,和不同的基因組特徵(genomic feature)。通常由長條狀的展示框(track)組成,以基因序列的位置作為線性軸,目前並沒有一套標準用來展示各式各樣的基因組特徵,所以頗需要一定的經驗才能上手。
通常基因組瀏覽器可以用來視覺化fasta、bed、gtf/gff、sam/bam檔,且大多數的基因組瀏覽器都有預先放置人類和老鼠的基因組參考序列。
最陽春的方式來瀏覽bam檔可以使用samtools tview的功能,但需要較複雜的探索性分析還是需要基因組瀏覽器的幫助,下面是目前還在active維護的基因組瀏覽器,大多都具備相當不錯的表現能力,幾乎都以java寫的,所以在安裝的時候要注意java相關的依賴,在啟動的時候大多都可以調整記憶體需求,也能利用remote的方式傳送影像。
IGV
上次更新時間:2016/11/1

由Broad institue所開發,元老級的基因瀏覽器,是目前“市佔率”最大的基因組瀏覽器,在2013年的時候性能有一次大升級。
Artemis
上次更新時間:2017/1/31
由wellcome trust sanger institue所開發,可同時組裡genebank和ensemb格式,且其能手動去調整註釋,另外,也是ensembl內部在組裡資料所使用的軟體
SeqMonk
上次更新時間:2017/6/2
由開發fastqc軟體的Babraham institue所開發,對於許多chipseq, methylation等3d structure資料有支持
iobio
上次更新時間:2017/7

唯一提供線上基因組資料瀏覽和分析的工具,整體軟體使用起來很舒服,為University of Utah研究團隊Marth lab所開發的軟體,在展示variant時,還會提供各種variant score來做為搜尋disease-causing variant幫助使用
IGB
上次更新時間:2016/6/7
在2001就被Affymetrix開發出來了,直到現在都還在維持中,他的使用手冊是這幾個genome browser中算是寫得最詳盡和清楚的,可以用來瀏覽Chipseq、RNAseq等等輸出資料
tablet
上次更新時間:2016/9/6

由The James Hutton Institue所開發的瀏覽器


 Illumina sequence identifiers
Illumina sequence identifiers

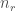 代表單一個gene其上的reads數量,L代表是這段基因的總長度除1000(轉換成kilobase的單位),N則是總共的reads數,通常用於singel reads
代表單一個gene其上的reads數量,L代表是這段基因的總長度除1000(轉換成kilobase的單位),N則是總共的reads數,通常用於singel reads


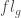 則代表是這基因的exon總長度
則代表是這基因的exon總長度
 qstat:用來看目前任務的運行狀況和單個任務所使用的資源
qstat:用來看目前任務的運行狀況和單個任務所使用的資源