reference:
Trapnell, C., Williams, B. a, Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., … Pachter, L. (2010).,Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28(5), 511–515. http://doi.org/10.1038/nbt.1621
cufflinks是一款用c++撰寫的程式,可以用來處理RNAseq已經align玩的資料,將其alignmenta組合成transcipts,並且找尋不同isoforms的可能,操作上在command line下使用。可以在這邊的網站下載以及使用說明書。
原理
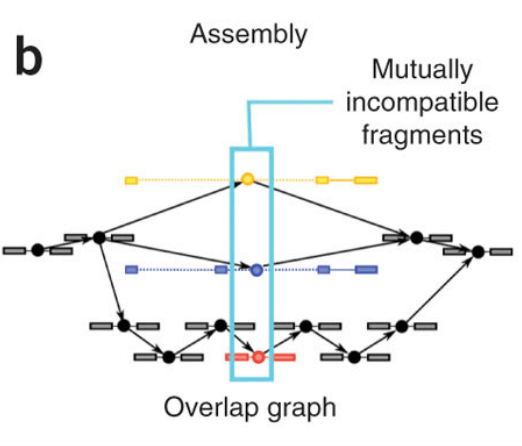
第一步:在paired-end的設計下,其會把一對的reads一起看,將其排列成overlapped的bundles,程式會先找出incompatible的fragment,因為這些特別的點代表者一種spliced isoforms的可能,如下圖所示,黃色藍色和紅色分別都是被標示的incompatible fragments,整個資訊會用node和edge的方式串接起來
第二步,會使用這種graph產生出isoforms的可能組合

軟體使用的注意事項:
- 假如是paired-end reads實驗設計,要把reads identified的數值要改成一樣的。
- samtools必須安裝好,且路徑設計在PATH變數下
語法:

–overlap–radius:default 50 base-pair
這是用來找出比較長距離的gene models
-g reference.gtf
可以給定已知的gene model reference
輸出:
總共會輸出四個檔案:transcripts.gtf isoforms.fpkm_tracking genes.fpkm_tracking
小建議:
假如要有多組sample要做cufflinks的話,建議先把bam檔合成單一個檔案,因為當alignment數越多,使用其演算法能找出的獨特isoforms較多!
