原始論文:Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D., & Craig, D. W. (2016). Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature Reviews Genetics, 17(5), 257–271. doi:10.1038/nrg.2016.10
論文的最後,談到這些新科技如何整合進臨床之中
第一個重點就是“新的檢測(這邊指得當然就是RNAseq)”,要能變成現行臨床工作的一部份,需要符合許多特性,可以由三個層面一步步來看,分別是Analytical validity (檢測本身的效力)、Clinical validity(臨床上的檢測能力)、Clinical utility(對於最終的臨床決策的貢獻度)。
第一步:Analytical validity
相對於實驗室裡的檢測,處理的檢體都是在control condition下,所以要能讓每一次的實驗都能reproducible的技術門檻就比較低,對比於臨床簡體的複雜性,還要能在此狀況下維持檢驗的穩定性、感測極限等等,就是第一關的重要門檻,要能在有良好的檢測標準來提供檢測sensitivity、specificity的驗證,再來是每次重複的結果應該要相似的(reproducibility),不會因為微小的變數造成數值的大波動。(robustness),如前exosome的研究便是因為其容易波動而造成無法定量。
以RNAseq來看,撇開前製定序所花的不算,光後端的分析方式、參數的差異,就會造成結果有所不同,在2013年,Genetic European Variant in Disease consortium為了解決這問題設計了一組實驗來處理這些technical reproducibility和feasible的問題,收集了465個人的lymphoblastoid cell,在七個定序中心分別執行,最後總結了一些在後端上可以用來驗證定序結果的,像是GC content、fragment size、transcript length、percentage of reads mapped to annotated exon (這部分可以參考所謂的annotation QC)。除此之外,由FDA領頭的SEQC study和ABRF study也有大量研究為了解決和提出相關標準。
Su, Z. et al. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 32, 903–914 (2014).
Li, S. et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol. 32, 915–925 (2014).
第二步: Clinical validity
當檢測技術能在臨床檢體上保持者可重複性時,接下來檢測就要達成可以實際區分biological difference的區別力。
第三步: Clinical utility
當前兩部都達成後,再來要看這個檢驗的結果能如何改變治療,如同companion diagnostic等等,通常此時就需要setup 臨床的試驗,來看實際在臨床場域中的效果!
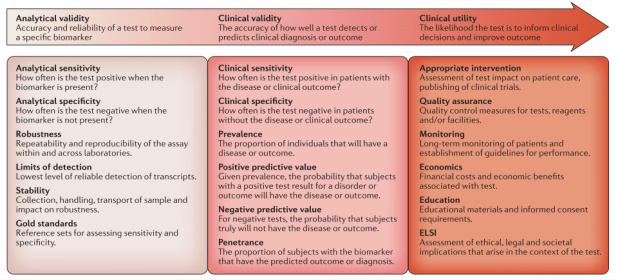
除此之外,FDA也有針對RNAseq為主的檢測項目做出一些法規的架構,可以由此深入看美國FDA對於NGS技術延生的檢測所抱持的想法
US Department of Health & Human Services. Center for Devices and Radiological Health. FDA notification and medical device reporting for laboratory developed tests (LDTs) — draft guidance. [online] (2014).
US Department of Health & Human Services. Center for Devices and Radiological Health. Framework for regulatory oversight of laboratory developed tests (LDTs) — draft guidance. [online] (2014)
US Department of Health & Human Services. Optimizing FDA’ s regulatory oversight of next generation sequencing diagnostic tests — preliminary discussion paper. [online] (2014).
Evans, B. J., Burke, W. & Jarvik, G. P. The FDA and genomic tests—getting regulation right. N. Engl. J. Med. 372, 2258–2264 (2015).