Han Y, Gao S, Muegge K, Zhang W, Zhou B. (2015) Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights 9(Suppl 1):29-46. [article]
RNAseq的資料為high resolution到single base,可應用範圍寬廣
RNAseq的技術可以用來區分許多不同種類的transcripts像是long non-coding RNA、miRNA、siRNA、snRNA、piRNA,以及他們如何調控蛋白質的translation或是調控chromatin states
- Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
- Robertson G, Schein J, Chiu R, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7(11):909–12.
RNAseq也可以用來研究enhancer region的transcripts表現量,這可能跟epigenetic gene regulation相關,可以稱這類型的transcripts為enhancer RNA。
- Andersson R, Gebhard C, Miguel-Escalada I, et al; FANTOM Consortium. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507(7493):455–61.
- Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–7
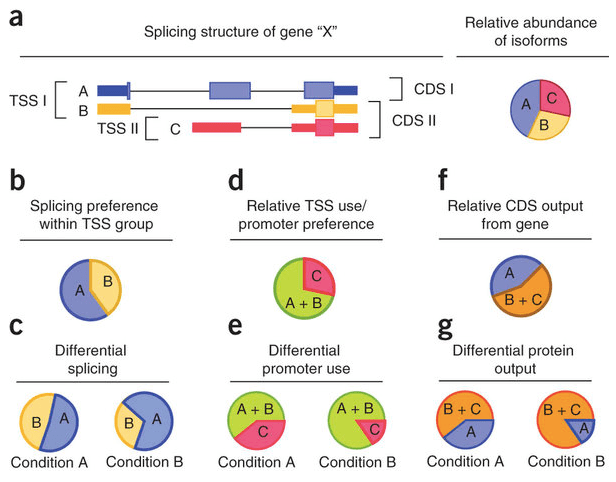
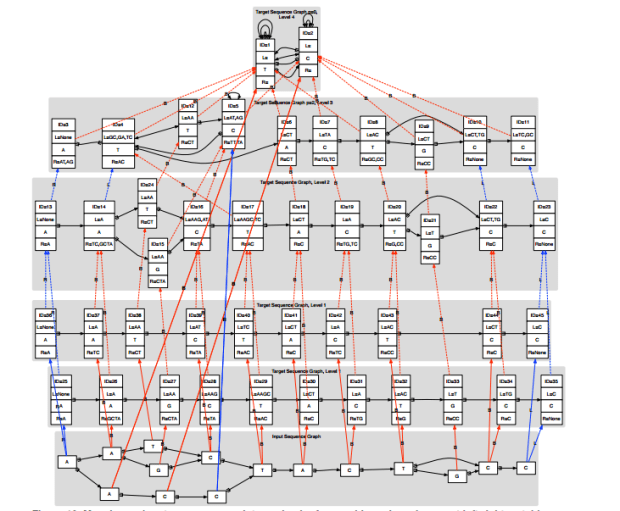
從RNAseq的資料可以取得transciption start sites、alternative promoter usage、mRNA isoforms (alternative splicing)、mRNA 穩定性(premature transciption termination at the 3′ )
- Camarena L, Bruno V, Euskirchen G, Poggio S, Snyder M. Molecular mecha- nisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pat- hog. 2010;6(4):e1000834.
- Griffith M, Griffith OL, Mwenifumbo J, et al. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7(10):843–7.
- Picardi E, Horner DS, Chiara M, Schiavon R, Valle G, Pesole G. Large- scale detection and analysis of RNA editing in grape mtDNA by RNA deep- sequencing. Nucleic Acids Res. 2010;38(14):4755–67.
- Wilhelm BT, Briau M, Austin P, et al. RNA-seq analysis of 2 closely related leukemia clones that differ in their self-renewal capacity. Blood. 2011;117(2): e27–38.
- Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6
Locating regulatory element
- Liu Y, Han D, Han Y, et al. Ab initio identification of transcription start sites in the Rhesus macaque genome by histone modification and RNA-Seq. Nucleic Acids Res. 2011;39(4):1408–18.
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013; 339(6123):1074–7.
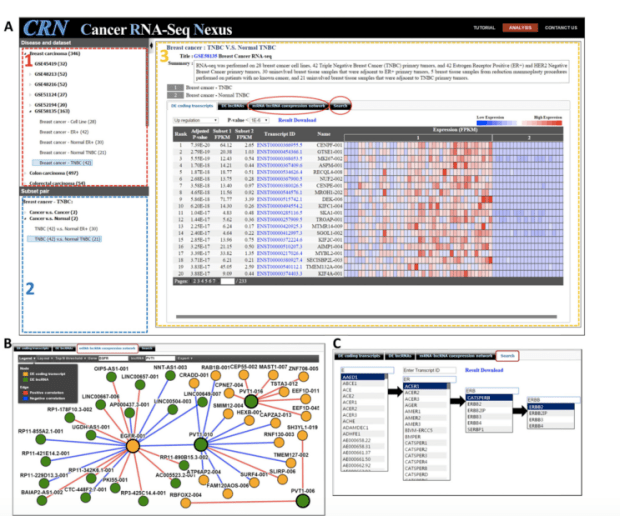
Allele-specific expression、disease-associated single nucleotide polymorphisms(SNP)、fusion gene for causal variants in cancer
- Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458(7234):97–101.
- Berger MF, Levin JZ, Vijayendran K, et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20(4):413–27.
- Supper J, Gugenmus C, Wollnik J, et al. Detecting and visualizing gene fusions. Methods.2013;59(1):S24–8
- Conde L, Bracci PM, Richardson R, Montgomery SB, Skibola CF. Integrating GWAS and expression data for functional characterization of disease-associated SNPs: an application to follicular lymphoma. Am J Hum Genet. 2013;92(1):126–30.
Transcription of endogenous retrotransposons and parasitic repeat elements that influence the transcription of neighboring genes or result in somatic mosaicism in the brain
- Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15(8):497–506.