# Shell 說明
#!/bin/bash
# Slurm 指令
#SBATCH -A ACD110078 # Account name/project number
#SBATCH -J hello_world # Job name
#SBATCH -p test # Partiotion name
#SBATCH -n 24 # Number of MPI tasks (i.e. processes)
#SBATCH -c 1 # Number of cores per MPI task
#SBATCH -N 3 # Maximum number of nodes to be allocated
#SBATCH -o %j.out # Path to the standard output file
#SBATCH -e %j.err # Path to the standard error ouput file
#程式與指令
module load compiler/intel/2020u4 IntelMPI/2020
mpiexec.hydra -bootstrap slurm -n 24 /home/user/bin/intel-hello
Important things are going to happen. Synthetic biology could be increasingly important in environmental clean-up strategies. It could also have a place in planet colonization. If it happens, then planet colonization will be reliant on synthetic biology. The words ‘insurmountable opportunities’ come to mind. Asking that question is like asking Bardeen in 1948 after developing the transistor to ‘please predict iPhones’. You can’t predict it. You can make guesses about what’s important today but we don’t have a clue for the future .
Quote from Tom Knight
Ginkgo Biowork是一間在合成生物學商業領域很特別的公司,常被稱為波士頓生技獨角獸,這邊是閱讀Ginkgo Biowork : The Organism Company 這邊文章所摘錄的心得,這篇文章是由Elliot Hershberg所撰寫,發表在Not Boring上面,Not Boring是一間每週寄兩封電子信,分享關於Web3和商業內容,很驚訝他們也會分享如合成生物學領域的資訊,另外,這篇文章也提到一Ginkgo Bioworks很神奇的角度和領域。
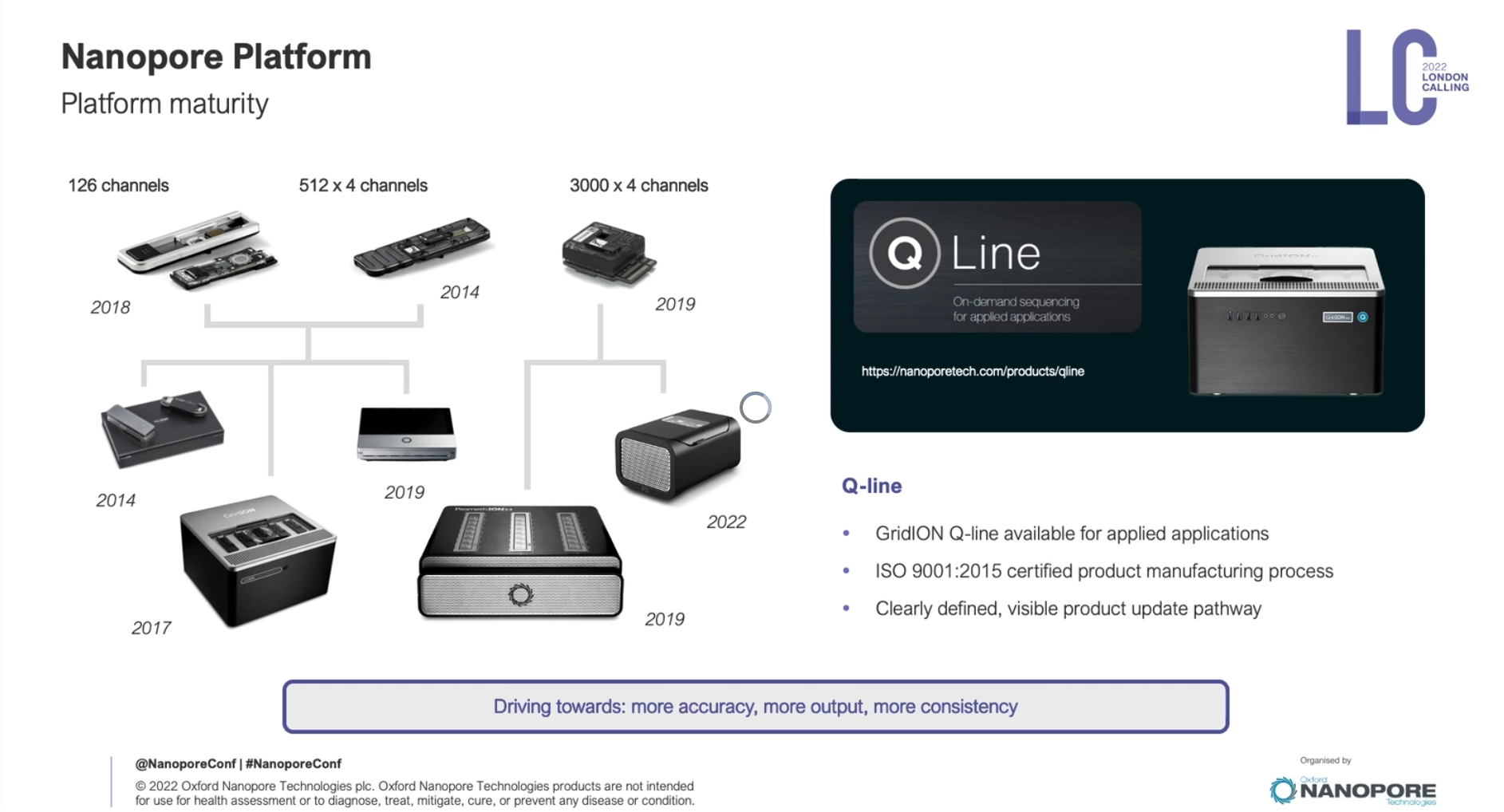
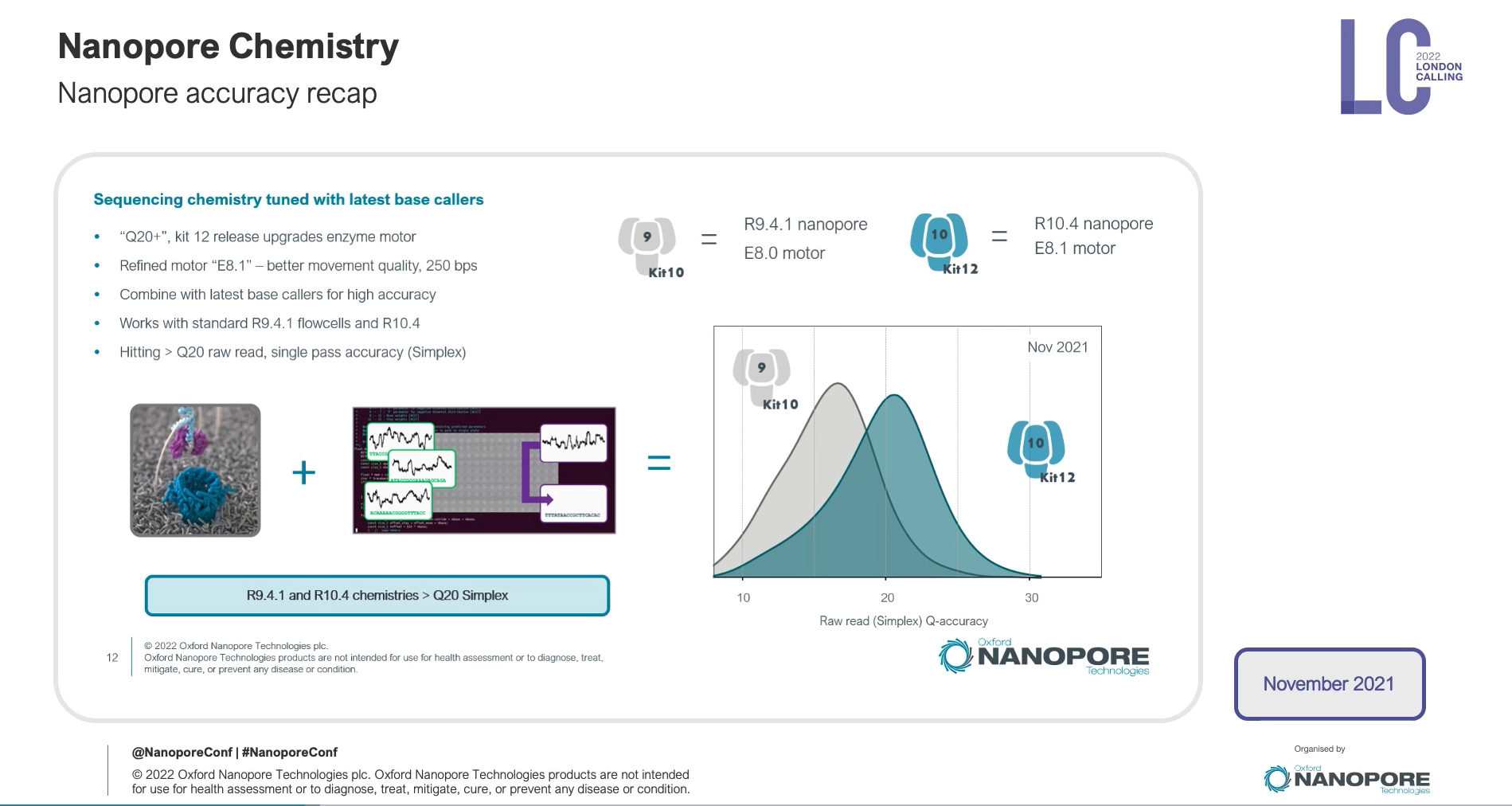
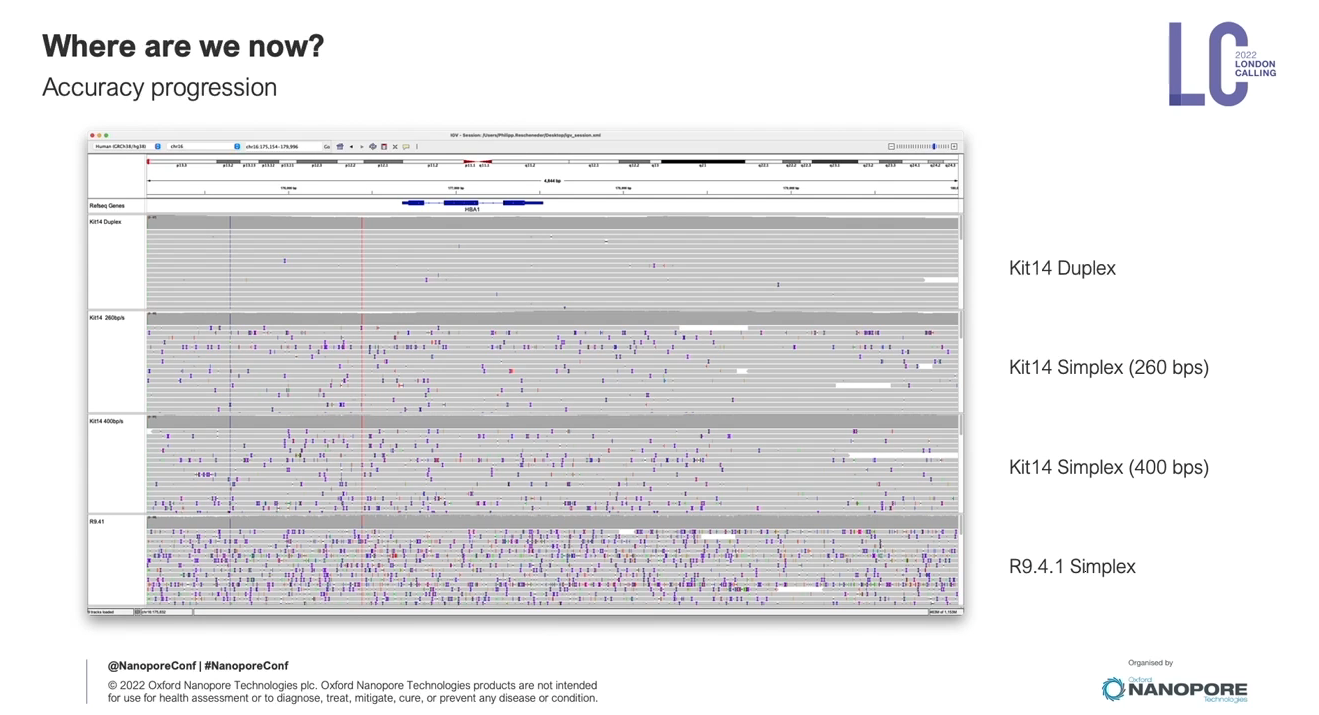
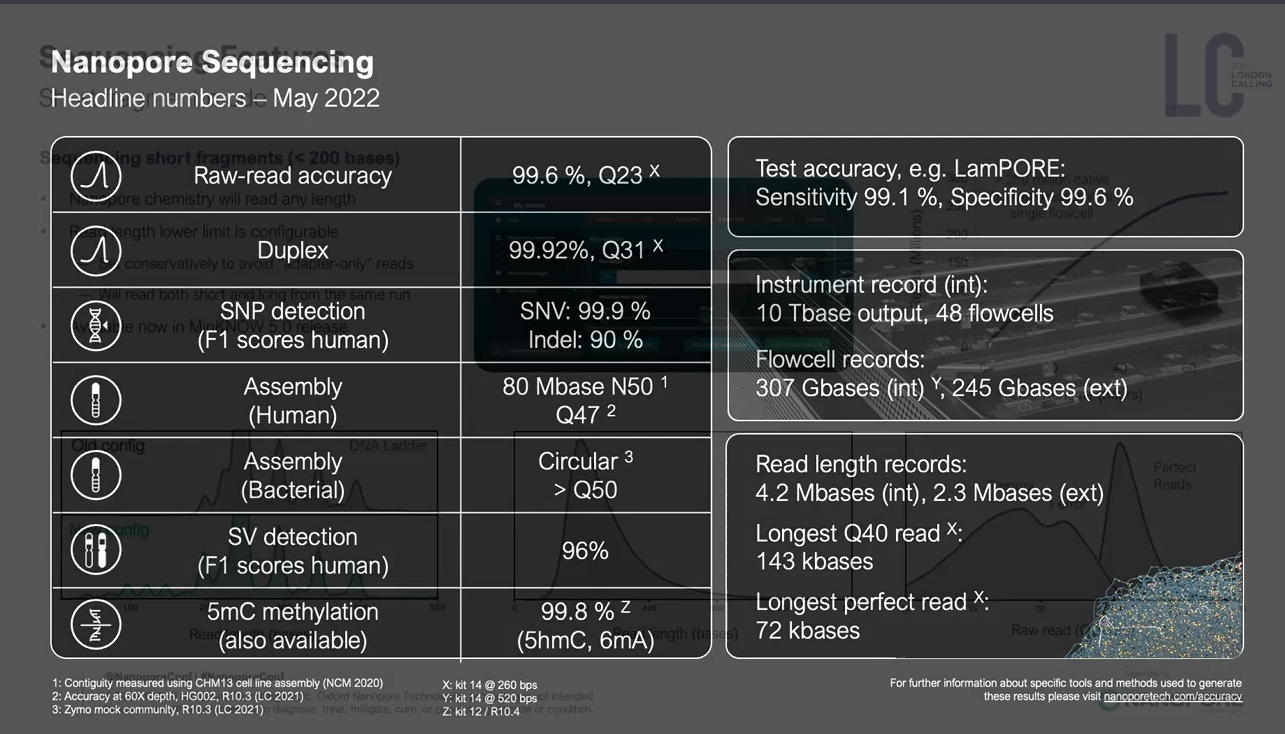
每年Oxford nanopore都會舉辦一場在總部London的會議,會議中會去介紹最新發展,這會議叫做London Calling,今年2022 London Calling,在五月中舉辦,實體和線上並行,對於暫時無法出國的人來說,是非常棒的,可以直接參與線上演講活動。
這場會議中,會邀請這一年有利用oxford nanoporepore的學者分享他們的經驗,也會邀請領域專家來給一些比較廣泛的主題,另外,最核心的一場演講是Update from Oxford Nanopore Technologies,由Oxford nanopore公司技術長Clive Brown 主持,其中各部門主管會來分享部門近況和更新,算是想要理解目前Oxford nanopore最新的發展,可以只聽這一場。這邊分享一下今年的一些蠻不錯的內容,可以感受到這個技術線的可擴展性,非常令人振奮!
Reading package lists... Done<br>Building dependency tree... Done<br>Reading state information... Done<br>Note, selecting 'rstudio' instead of './rstudio-2022.02.0-443-amd64.deb'<br>Some packages could not be installed. This may mean that you have<br>requested an impossible situation or if you are using the unstable<br>distribution that some required packages have not yet been created<br>or been moved out of Incoming.<br>The following information may help to resolve the situation:<
The following packages have unmet dependencies:<br> rstudio : Depends: libssl1.0.0 but it is not installable or<br> libssl1.0.2 but it is not installable or<br> libssl1.1 but it is not installable<br>E: Unable to correct problems, you have held broken packages.
General Design Procedure for Free and Open-Source Hardware for Scientific Equipment. 2018. Designs
A 3D Printer in the lab: Not only a toy, Vittorio Saggiomo
A Scientist’s Guide to Buying a 3D Printer: How to Choose the Right Printer for Your Laboratory
其實老爸拿到3D列印機後,便開始幫忙家裡很多零件的改造,也覺得以老爸的文筆可以好好結交世界的網友,這邊在老爸部落格建立前,先幫忙分享,此篇就是老爸閱讀由芬蘭瓦赫寧恩大學 (Wageningen University & Research)的學者Vittorio Saggiomo撰寫之文章A 3D Printer in the lab: Not only a toy,閱讀後的中文轉換,這篇文章也是在Twitter上所發現的。
實驗室中的 3D 列印機:不僅僅是玩具
“我們為什麼要購買 3D 列印機?
不幸的是,很多學生在要求為實驗室購買 3D 列印機時仍然從教授那裡得到這個問題
儘管 3D 列印機在家庭中變得越來越普遍,但它們在許多實驗室中的代表性仍然不足,在世界範圍內被視為玩具而不是實驗室設備。這篇簡短的評論想要改變這個保守的觀點看法。 如果你是一個得到“為什麼”的學生 問題,發送給你的教授的這個小評論。 如果你是 PI(Principal Investigator),尚未決定實驗室是否需要購買 3D 列印機,請務必閱讀這篇小評論,或者,如果你沒有時間閱讀,就買一台吧!將來你會感謝我的。
這篇小評論的重點是熔融沈積建模(FDM) 列印機以及獲得第一台3D列印機後會發生什麼事。簡而言之,這些列印機將塑料絲熔化並沉積逐層創建最終產品。 他們越來越更便宜,更容易使用,現在不難找到低於 500€歐元 的優質 3D 列印機。 以這樣的價格,即使不是最多功能的設備你應在實驗室裡裝一台3D 列印機。所以,你買了你的第一台 3D 列印機,組裝了它,現在它在實驗室裡。[1] 下一步是什麼?擁有一台 3D 列印機作為科學技術儀器有四個階段
:印刷、設計、材料和自動化。在以下段落中,我將詳細說明每個階段
(圖1)
Fig. 1. This mini-review focuses on the four phases of owning an FDM 3D printer: printing, designing, materials, and automation.
圖 1. 這篇小評論側重於擁有 FDM 3D 列印機的四個階段:列印、設計、材料和自動化。
第一步:進行列印
在最初的幾週內,幾乎可以肯定的是,列印機將用於列印大量玩具、小雕像和隨機物品。 這是一個正常的學習過程。 在此期間,用戶將了解 3D 列印機的工作原理及其詞典、切片 3D 設計、懸垂、橋接、邊緣、裙邊、填充幾何形狀、列印速度和溫度、回縮設置、如何列印不同材料等等。 這是一個必不可少的學習過程; 會有很多失敗,每一個都是寶貴的學習點。
圖 2. 3D 列印設計示例: a) 用於各種實驗室器具的模塊化支架:NMR 管、比色皿、獵鷹管,可從 [15] 下載。 b) 化學模型也可以從 [4] 下載和列印。 c) X 射線結構 [10]d) NMR 2D 光譜也可以轉換為 3D 可列印模型。 [11]
一旦隨機繪畫完成並且用戶熟悉了 3D 列印過程,此階段的第二階段是開始為實驗室列印有價值的物品(圖 2)。
在這裡,可能性是無止盡的。 最有可能的是,任何實驗室 3D 列印的第一個有用的東西是用於實驗室中每個小瓶、試管和比色皿的支架。[2,3] 有很多設計,從 NMR 支架到磁性 Eppendorf 支架 用於磁分離,可從 Thingiverse、Printables 或 NIH 3D 列印存儲庫等存儲庫在線免費獲取。 如果您需要某物的支架,它可能已經由其他人設計,並且可以下載和列印(圖 2a)。 即使您在此處停止閱讀,並且您的列印機僅用於列印支架,考慮到樣品支架的成本,您將在幾個月內獲得投資回報。
但是,一旦您將沈迷於3D 列印中,您會想要更多。 此時掃描文獻將返回大量可 3D 列印的對象:分子模型、[4,5,6] 晶體結構,這些都是有價值的教學模型,並為學生提供化學結構的物理模型等(圖 2b) . 例如,有關於如何將晶體學數據轉換為 3D 可列印模型的說明(圖 2c)[7-10] 甚至如何將 NMR 光譜轉換為 3D 列印(圖 2d)。 [11,12] 3D 列印和 NMR 不要在這裡結束:您可以列印用於固態 NMR [13] 甚至整個 NMR 磁體的樣品管。 [14] 修復破損的塑料實驗室物品也變得非常容易,許多移液器支架或移液器部件、連接器和桌面組織器都可以從存儲庫免費下載。
在某個時刻,您會意識到您需要的部件無法在線獲得,您的實驗室將進入擁有 3D 列印機的第二階段:設計。
對於科學家來說,至少擁有 3D 設計的基本知識,我怎麼強調都不為過,這不僅對於 3D 列印,而且對於為論文和演示文稿製作有吸引力的圖形也是如此。 學習 3D 設計可能看起來是一項艱鉅且不可能完成的任務,但相信我,這並不難,至少對於簡單的設計而言。 如果你能在紙上畫草圖,你就可以在電腦上設計它。
通過設計模型,您可以釋放 3D 列印的全部潛力。 現在,您可以生產非商業化的零件,它們非常適合您的應用或實驗室,或者是那裡其他儀器的替換或增強零件。 這就是快速原型製作的真正力量; 您可以在不到一天的時間內設計、列印、測試和重新設計一個對象,而無需從專門的在線服務訂購部件——這需要數週時間。 從頭開始創建一些東西並在幾個小時後掌握它是一種令人難以置信的體驗。
兩個科學領域極大地受益於 3D 列印的出現和直接在實驗室中進行快速原型製作的可能性:微流體[16,17] 和顯微鏡[18]。
圖 3. 3D 設計模型示例。 a) 一套微流體裝置和可使用各種塑料材料列印的連接器。 [27] b) 製造方法使用 3D 列印可溶解材料的 PDMS 微流體裝置。 [28] c) Openflex ure 顯微鏡,一種完全可 3D 列印的顯微鏡。 [32] d) 3D 列印的橫截面用於化學反應的實驗室器具。 [39] 一個解決方案被移動到不同的立方體中旋轉結構; 橙色和黃色兩種不同的催化劑直接 3D印在結構上
在 3D 列印出現之前,製造微流控設備是一項繁瑣的任務:其步驟包括使用軟光刻技術製作母版、成型和脫模 PDMS(聚二甲基矽氧烷)、多層微流控設備的對齊以及 PDMS 與玻璃的最終等離子鍵合。如果微流體未按預期工作,則該過程將重複多次以微調微流體參數。這個過程隨著 3D 列印發生了巨大的變化,各種設計可以在幾個小時內完成並列印出來,從而加快了研究速度並降低了原型製作成本。一種方法是在 3D 可列印塑料中設計和列印整個微流體裝置。[19-22] 列印單個塑料塊而不是混合 PDMS/玻璃裝置會大大增加其耐壓性。[23]此外,在列印過程中可以暫停列印過程,以在列印對象內添加其他對象,從而為設備添加新功能,[24] 這一過程在標準微流體製造中通常是不可能的。例如,該過程已被用於在 3D 列印微流體設備中直接添加 PMMA 觀察窗口 [25] 甚至電化學檢測器。 [26]還有一個用於微流體的 3D 列印模型的開源存儲庫,不僅包括設備本身,還包括所有連接和 Luer 適配器(圖 3a)。 [27]
使用 3D 列印機製造微流控設備的另一種選擇是利用一些 3D 列印塑料的溶解度來發揮我們的優勢。 2015年,我們團隊列印ABS並將其嵌入到PDMS中。 當將此 PDMS/ABS 塊置於丙酮中時,ABS 會溶解,在 PDMS 內部留下一個開放通道(圖 3b)。 [28] 該方法可以製作單塊 PDMS 微流控設備,這是微流控領域的標準且特性非常好的材料,僅使用標準 3D 列印機和 3D 列印材料即可,無需昂貴或難以使用的儀器。 該方法已應用於水溶性材料,[29] 或用於製造互連的 PDMS/玻璃微流體塊。 [30] 3D 列印也被用於製造紙質微流體,使用 3D 列印材料作為濾紙上液體的屏障。 [31]
另一個從 3D 列印中受益匪淺的領域是顯微鏡。快速且廉價地製作顯微鏡零件原型的可能性使該領域民主化,並開啟了製造全 3D 列印顯微鏡的可能性。最常見的 3D 列印顯微鏡可能是 OpenFlexure(圖 3c)。 [32]它使用 3D 列印平台來實現幾微米的穩定運動。在這種彎曲的基礎上,可以 3D 列印亞 100nm 光纖對準台。 [33]如果您認為舞台是一塊 3D 列印的塑料,這令人印象深刻。 3D 列印機的使用也為自製顯微鏡的製造打開了大門,其中各種 3D 列印光學塊可以組合在一起用於不同的顯微鏡設置。 µCube[34] 是這種方法的第一個例子,後來 UC2(你也看到了)改進了這個過程,消除了對塊的繁瑣對齊的需要。 [35]只需在這些設置上重新調整立方體即可在幾分鐘內將明場顯微鏡轉變為熒光顯微鏡。此外,3D 列印還被用於製造單分子顯微鏡:miCube。 [36]可以在此處找到開放式顯微鏡數據庫 [37]。
如果我告訴你,你甚至可以設計你的化學反應器呢? 這已被克羅寧的團隊多次證明。 在列印過程中添加化學品和外部組件(如過濾器或矽膠)的可能性允許用戶在幾個小時內完成整個合成和純化設置。 3D 列印的多功能性以及在 3D 列印物體中插入外部組件的可能性允許精確且可重複的合成和純化步驟。 Cronin 的小組通過列印具有不同功能的立方體證明了這一點,並且通過簡單地轉動 3D 列印物體將溶液從一個塊移動到另一個塊來執行單個合成和純化步驟(圖 3d)。[38-40] 該小組還 事實證明,使用聚丙烯 (PP) 作為 3D 列印材料,可以使容器在高溫和高壓下保持穩定。 [41]
3D 列印還用於分析化學,這是另一個廣泛使用 3D 列印多功能性來設計和列印分離裝置、流動池、混合器、濃縮器等的領域。 [42-45]
如果您打算將材料用於您感興趣的解決方案,請注意有關材料的警告。市售的 3D 列印材料很少或從來不是純聚合物。 PLA、PETG、PP等,不是100%純;它們總是含有一些共聚物、添加劑,在有色材料的情況下,還含有某種顏料。 3D 列印材料的配方使其易於列印,並且具有機械性能,而不是純度。即使是來自同一供應商的相同材料,批次之間的化學成分也可能不同。不要假設這些材料是純聚合物,如果您打算將它們與您感興趣的溶液接觸,例如用於合成或分析目的,請務必提前測試材料。這可以通過溶解材料並記錄 NMR 來完成,或者更快地將 3D 列印材料放入您計劃使用的溶劑中,讓它在那裡放置幾天,然後記錄溶液的 UVVis 光譜,尋找可能的分子洩漏。兩者都是快速的方法,至少可以為您提供成分的指示以及在實驗過程中從材料中洩漏的可能性。
同樣在這個階段,你會聽到一個經典的批評:“3D 設計太難了”。我相信你在學習過程中遇到過微積分、熱力學和量子力學,並從中倖存下來。與這些主題相比,3D 設計是在公園裡散步。如今,有很多易於使用的 3D 設計軟件,最簡單的是 Tinkercad,一種免費的在線(獨立於平台)設計。在 10 分鐘關於如何使用它的 YouTube 教程之後,我可以向您保證,您將能夠在幾分鐘內製作出您的第一個 3D 設計。對於更複雜的設計,最常用的軟件是 Solidworks、Fusion 360 和 Blender。前兩個是商業的,而第三個是開放軟件。它們使用起來更複雜,需要更多的培訓,但沒有什麼是不可能學習的。如果您喜歡編程,另一種選擇是 OpenSCAD,它使用類似編程的結構和語法來設計 3D 模型。 Shapr3D 是最新的軟件,最初是為 iPad+Stylus 設計的,但現在可用於 Mac 和 PC,它對學術界免費且使用非常直觀。再一次,3D 設計不僅對於 3D 列印,而且對於製作有吸引力的科學方案和圖形也是一項有用的技能。
第三階段:材料
您已經掌握了 3D 列印並正在習慣 3D 設計。 下一步是什麼? 顯然:製作自己的材料。在 FDM 中,這意味著將塑料溶解在適當的溶劑中,添加感興趣的外部材料,蒸發溶劑,撕碎塑料複合材料,然後將其擠出至使用諸如 FelFil Evo、Filabot、3Devo 等商用長絲擠出機或通過構建您的擁有自己的長絲擠出機,例如 Lyman Extruder V5。 這個過程並不容易,市售的燈絲擠出機比 3D 列印機貴,因為較小的細分市場。 但是,您可以製作自己的複合材料或多重材料,並且列印成任何你想要的形狀,這在十年前是不可能的想像。 [46-48]
但您為什麼要製作用於 3D 列印的複合材料? 利用這一過程的主要領域之一是製藥領域,使用特定的配方 [49-52] 和 3D 列印個性化片劑和定制的藥物釋放曲線(圖 4a)。 [53-55] 也已經完成了研究 檢查復合藥用長絲的可印刷性。 [56] 在醫學領域,正在開發的複合 3D 可列印材料主要用於抗菌和醫療級材料,用於鑄件、假肢和可包覆器官的物體[57-62]
圖 4. 不同材料的示例以及 3D 列印在製藥中的影響,其中可以隨意改變藥丸的結構以改變其溶解度時間(一)。 類似的方法可用於 3D 列印催化劑,其中結構可以改變以具有或多或少的表面積和存在的催化劑 (b)。一種可以模仿 Lycurgus 杯的納米複合材料,顯示出兩種不同的反射(綠色)和透射(紅色)中的顏色(c)。 [100]
在 3D 列印燈絲中插入外部化學物質的可能性使我們能夠列印增強對象。 這可以通過機械性能來實現,例如,通過使用天然填料,甚至大麻,[63] 來修改列印對象的機械性能。 [64] 金屬、[65] 木材、[66] 和石墨[67] 也已被用於改變 3D 列印物體的機械性能。 但是,對於機械性能而言,這種方法最極端的情況可能是在擠出過程中嵌入連續纖維。 [68] 在這種情況下,連續纖維被插入並驅動到噴嘴內,同時它沉積熔融塑料,從而將連續線嵌入每條沉積的塑料線中,從而大大提高最終 3D 列印部件的機械強度
本段只是對製作您自己的 3D 列印複合材料的多種可能性的簡短介紹,並不意味著詳盡無遺,因為複合 3D 列印材料還有其他領域會大放異彩。 然而,我認為有必要展示至少一些機會,這些機會不僅來自於設計具有特定特徵的對象,例如具有高表面積或混合能力,而且來自於將它們與獨特的、應用程序驅動的、複合材料相結合。 實驗室製作的材料
這裡有一點警告是關於 FDM 列印方法的。 由於燈絲需要熔化才能沉積在 3D 列印床上,因此根據材料,在 3D 列印機熱端將其加熱到 180°C 以上。 這也意味著您添加到塑料中的每種化合物和材料也會經受高溫,即使時間很短。 這種高溫可能是有害的,尤其是對有機分子,列印這些複合材料時應該考慮到這一點。 一種監測列印過程中有機分子降解的方法是在 3D 列印過程後溶解材料,並使用 NMR、MS 和 UV-Vis 重新表徵有機化合物。
如果您想製作複合材料並在不列印的情況下列印 經受高溫,您可能需要研究直接墨水書寫 (DIW) 3D 列印。 這種3D列印方法使用注射器以逐層方式沉積糊劑,類似於 FDM。立體光刻 (SLA) 3D 列印機,有時稱為“樹脂”列印機,也被視為非高溫列印機。 然而,這是誤導性的,因為甲基丙烯酸酯光聚合反應是放熱的,並且局部容易達到超過 100°C。
階段4:自動化
3D 列印機是 3 軸機器人,帶有一個額外的電機來推動燈絲通過噴嘴,由於消費市場,它們一天比一天便宜,而且有可能不到 200 攝氏度的價格購買 3D 列印機。如果您考慮它,這些是市場上最實惠的三軸機器人。
舉個極端的例子,如果你買了一台 3D 列印機,用它不是用來列印,而是用來來回移動列印床使用試管、液體細胞培養物等,它更便宜比您可以購買的任何振動器/搖桿。 3D的組成部分列印機可分為機械部分和電氣部分組件。所有的 3D 列印機都有三個線性運動系統。這些可以是皮帶、導軌或絲槓,具體取決於該模型。它們至少有四個步進電機、兩個加熱元件、兩個控制溫度的熱敏電阻、三個觸摸開關(終點站),大量的螺釘、螺母、螺栓和組裝它們的所有必要工具。在電子部分,他們有一個主板、一個電源單元和所有的用於將主板連接到其他組件的電纜。 3D列印消費市場增長如此之快,如果您嘗試購買所有單個零件比購買 3D 列印機。
如果我們沒有辦法,所有這些好東西都將毫無用處編程和控制“機器人”的運動。 這個可以通過直接利用 3D 列印機編程來完成語言。 市場上幾乎所有的列印機都使用開源Marlin 固件和 G-Code 作為編程語言。
為了使 3D 列印機成為實驗室使用的標准設備,我們可以改進以下幾點: 適當的數據庫仍然缺乏。到目前為止,設計在不同的數據庫,呈現對特定設計的搜索麻煩。 幾年前NIH 製作了一個 3D 列印零件數據庫,但許多研究人員更喜歡使用其他的。這是維護得不好,而且幾乎找不到零件。為了解決這個問題,一個具有良好的統一數據庫搜索引擎和 DOI 或類似 DOI 的系統以正確引用該設計是理想的。還有一個很重要的就是教關於 3D 列印和 3D 設計。 3D 的基本原理列印、3D 設計和編程應該是幾乎所有科學家的課程。在這裡,我對您的建議是在您的課程中引入 3D 列印,而與您所教的內容無關,即使不到一個小時。新技術將嚴重影響下一代科學家並引入 3D 列印和編程盡快在他們的課程中加入將對學生的發展有很大的幫助。
要考慮的另一點是塑料廢物。當世界試圖減少塑料材料的使用時,3D 列印在“綠色”方面似乎違反直覺材料。然而,除了 3D 列印的綠色優勢一般來說,與行業標準相比,例如印刷僅一件,使用地點印刷,無需出貨等,綠色(er)材料開始出現在市場上。回收PETG或PLA目前可用來自許多不同的供應商。但是,我們可以做得更好,只使用回收材料。例如,支撐材料應盡可能避免。支撐材料是塑料材料,一旦列印完成即被扔掉,因此在可能的情況下,應注意對象的設計無需使用任何工具即可列印支持材料。對於多色或多材料列印機也可以提出類似的反對意見,因為大多數材料是在“彩色塔”中被清除。再一次,它是廢物,應盡可能避免。塑料垃圾來自 3D 列印的產品只是滄海一粟,它是全球塑料垃圾中極小的一部分。但是,關於該主題的討論以及如何避免不必要的浪費或
盡量減少它對於道德研究環境至關重要。
我希望我設法讓你相信實驗室裡的 3D 列印機。 這篇簡短的評論只關注在 FDM 列印機上,因為我相信這是第一步在 3D 列印中,是最通用的一種。 或早或晚稍後,您將遇到立體光固化成型 (SLA) 或蒙版立體光刻 (m)SLA 列印機。 他們使用混合甲基丙烯酸酯和光聚合反應製造 3D 物體。 如果一個學生要這種列印機,買它們,他們可能比你知道的更多(還有我,為了它的價值)。
相關文獻
1. Tully, J.J., and Meloni, G.N. (2020) A Scientist’s Guide to Buying a 3D Printer: How to Choose the Right Printer for Your Laboratory. Anal. Chem., 92 (22), 14853–14860.
2. Baden, T., Chagas, A.M., Gage, G., Marzullo, T., PrietoGodino, L.L., and Euler, T. (2015) Open Labware: 3-D Printing Your Own Lab Equipment. PLOS Biol., 13 (3), e1002086.
3. Lücking, T.H., Sambale, F., Schnaars, B., Bulnes-Abundis, D., Beutel, S., and Scheper, T. (2015) 3D-printed individual labware in biosciences by rapid prototyping: In vitro biocompatibility and applications for eukaryotic cell cultures. Eng. Life Sci., 15 (1), 57–64.
4. Penny, M.R., Cao, Z.J., Patel, B., Sil dos Santos, B., Asquith, C.R.M., Szulc, B.R., Rao, Z.X., Muwaffak, Z.,Malkinson, J.P., and Hilton, S.T. (2017) Three-Dimensional Printing of a Scalable Molecular Model and Orbital Kit for Organic Chemistry Teaching and Learning. J. Chem. Educ., 94 (9), 1265–1271.
5. Jones, O.A.H., and Spencer, M.J.S. (2018) A Simplified Method for the 3D Printing of Molecular Models for Chemical Education. J. Chem. Educ., 95 (1), 88–96.
6. Paukstelis, P.J. (2018) MolPrint3D: Enhanced 3D Printing of Ball-and-Stick Molecular Models. J. Chem. Educ., 95 (1), 169–172.
7. Chen, T.-H., Lee, S., Flood, A.H., and Miljanic, O.Š. ´ (2014) How to print a crystal structure model in 3D. CrystEngComm, 16 (25), 5488–5493.
8. Kitson, P.J., Macdonell, A., Tsuda, S., Zang, H., Long, D.-L., and Cronin, L. (2014) Bringing Crystal Structures to Reality by Three-Dimensional Printing. Cryst. Growth Des., 14 (6), 2720–2724.
9. J. Mithila, F., Oyola-Reynoso, S., M. Thuo, M., and B.J. Atkinson, M. (2016) Visualization of Hyperconjugation and Subsequent Structural Distortions through 3D Printing of Crystal Structures. Lett. Org. Chem., 13 (4), 272–276. 10. Brown, M.L., Hartling, D., Tailor, H.N., Van Wieren, K., Houghton, G.B., McGregor, I.G., Hansen, C.D., and Merbouh, N. (2019) Piecewise 3D printing of crystallographic data for post-printing construction. CrystEngComm, 21 (38), 5757–5766.
11. Bakker, M., Boyd, B., and Meints, G.A. (2018) 3D printed NMR spectra: From 1D and 2D acquisition to 3D visualization. Concepts Magn. Reson. Part A, 47A (1), e21470.
12. Jones, O.A.H., Stevenson, P.G., Hameka, S.C., Osborne, D.A., Taylor, P.D., and Spencer, M.J.S. (2021) Using 3D Printing to Visualize 2D Chromatograms and NMR Spectra for the Classroom. J. Chem. Educ., 98 (3), 1024–1030.
13. Long, Z., Ruthford, J., and Opella, S.J. (2021) 3D printed sample tubes for solid-state NMR experiments. J. Magn. Reson., 327, 106957.
14. Alnajjar, B.M.K., Buchau, A., Baumgärtner, L., and Anders, J. (2021) NMR magnets for portable applications using 3D printed materials. J. Magn. Reson., 326, 106934.
17. Mehta, V., and Rath, S.N. (2021) 3D printed microfluidic devices: a review focused on four fundamental manufacturing approaches and implications on the field of healthcare. Bio-Des. Manuf., 4 (2), 311–343.
18. Del Rosario, M., Heil, H.S., Mendes, A., Saggiomo, V., and Henriques, R. (2021) The Field Guide to 3D Printing in Optical Microscopy for Life Sciences. Adv. Biol., 2100994.
19. Li, J., Baxani, D.K., Jamieson, W.D., Xu, W., Rocha, V.G., Barrow, D.A., and Castell, O.K. (2020) Formation of Polarized, Functional Artificial Cells from Compartmentalized Droplet Networks and Nanomaterials, Using One-Step, Dual-Material 3D-Printed Microfluidics. Adv. Sci., 7 (1), 1901719. 20. Morgan, A.J.L., Hidalgo San Jose, L., Jamieson, W.D., Wymant, J.M., Song, B., Stephens, P., Barrow, D.A., and Castell, O.K. (2016) Simple and Versatile 3D Printed Microfluidics Using Fused Filament Fabrication. PLOS ONE, 11 (4), e0152023.
21. Tiboni, M., Benedetti, S., Skouras, A., Curzi, G., Perinelli, D.R., Palmieri, G.F., and Casettari, L. (2020) 3Dprinted microfluidic chip for the preparation of glycyrrhetinic acid-loaded ethanolic liposomes. Int. J. Pharm., 584, 119436. 22. Dragone, V., Sans, V., Rosnes, M.H., Kitson, P.J., and Cronin, L. (2013) 3D-printed devices for continuous-flow organic chemistry. Beilstein J. Org. Chem., 9, 951–959.
23. Romanov, V., Samuel, R., Chaharlang, M., Jafek, A.R., Frost, A., and Gale, B.K. (2018) FDM 3D Printing of HighPressure, Heat-Resistant, Transparent Microfluidic Devices. Anal. Chem., 90 (17), 10450–10456.
24. Yuen, P.K. (2016) Embedding objects during 3D printing to add new functionalities. Biomicrofluidics, 10 (4), 044104.
25. Bressan, L.P., Adamo, C.B., Quero, R.F., de Jesus, D.P., and da Silva, J.A.F. (2019) A simple procedure to produce FDM-based 3D-printed microfluidic devices with an integrated PMMA optical window. Anal. Methods, 11 (8), 1014–1020.
26. Carvalho, R.M., Ferreira, V.S., and Lucca, B.G. (2021) A novel all-3D-printed thread-based microfluidic device with an embedded electrochemical detector: first application in environmental analysis of nitrite. Anal. Methods, 13 (11), 1349–1357.
27. Price, A.J.N., Capel, A.J., Lee, R.J., Pradel, P., and Christie, S.D.R. (2021) An open source toolkit for 3D printed fluidics. J. Flow Chem., 11 (1), 37–51. 28. Saggiomo, V., and Velders, A.H. (2015) Simple 3D Printed Scaffold-Removal Method for the Fabrication of Intricate Microfluidic Devices. Adv. Sci., 2 (9), 1500125.
29. Dahlberg, T., Stangner, T., Zhang, H., Wiklund, K., Lundberg, P., Edman, L., and Andersson, M. (2018) 3D printed water-soluble scaffolds for rapid production of PDMS micro-fluidic flow chambers. Sci. Rep., 8 (1), 3372.
30. Felton, H., Hughes, R., and Diaz-Gaxiola, A. (2021) Negligible-cost microfluidic device fabrication using 3Dprinted interconnecting channel scaffolds. PLOS ONE, 16 (2), e0245206.
31. Zargaryan, A., Farhoudi, N., Haworth, G., Ashby, J.F., and Au, S.H. (2020) Hybrid 3D printed-paper microfluidics. Sci. Rep., 10 (1), 18379. 32. Sharkey, J.P., Foo, D.C.W., Kabla, A., Baumberg, J.J., and Bowman, R.W. (2016) A one-piece 3D printed flexure translation stage for open-source microscopy. Rev. Sci. Instrum., 87 (2), 025104.
33. Meng, Q., Harrington, K., Stirling, J., and Bowman, R. (2020) The OpenFlexure Block Stage: sub-100 nm fibre alignment with a monolithic plastic flexure stage. Opt. Express, 28 (4), 4763.
34. Delmans, M., and Haseloff, J. (2018) µCube: A Framework for 3D Printable Optomechanics. J. Open Hardw., 2 (1), 2.
35. Diederich, B., Lachmann, R., Carlstedt, S., Marsikova, B., Wang, H., Uwurukundo, X., Mosig, A.S., and Heintzmann, R. (2020) A versatile and customizable low-cost 3Dprinted open standard for microscopic imaging. Nat. Commun., 11 (1), 5979.
36. Martens, K.J.A., van Beljouw, S.P.B., van der Els, S., Vink, J.N.A., Baas, S., Vogelaar, G.A., Brouns, S.J.J., van Baarlen, P., Kleerebezem, M., and Hohlbein, J. (2019) Visualisation of dCas9 target search in vivo using an openmicroscopy framework. Nat. Commun., 10 (1), 3552.
37. Hohlbein, J., Diederich, B., Prakash, K., Shepherd, D., Jahr, W., Phoess, Smit, J., Tutkus, M., Haase, R., Moore, J., Barth, A., Vladimirov, N., and Bowman, R. (2022) HohlbeinLab/OpenMicroscopy: V01release,Zenodo.
38. Hou, W., Bubliauskas, A., Kitson, P.J., Francoia, J.- P., Powell-Davies, H., Gutierrez, J.M.P., Frei, P., Manzano, J.S., and Cronin, L. (2021) Automatic Generation of 3DPrinted Reactionware for Chemical Synthesis Digitization using ChemSCAD. ACS Cent. Sci., 7 (2), 212–218.
39. Kitson, P.J., Glatzel, S., Chen, W., Lin, C.-G., Song, Y.- F., and Cronin, L. (2016) 3D printing of versatile reactionware for chemical synthesis. Nat. Protoc., 11 (5), 920–936. 40. Kitson, P.J., Symes, M.D., Dragone, V., and Cronin, L. (2013) Combining 3D printing and liquid handling to produce user-friendly reactionware for chemical synthesis and purification. Chem Sci, 4 (8), 3099–3103.
41. Kitson, P.J., Marshall, R.J., Long, D., Forgan, R.S., and Cronin, L. (2014) 3D Printed High-Throughput Hydrothermal Reactionware for Discovery, Optimization, and ScaleUp. Angew. Chem. Int. Ed., 53 (47), 12723–12728.
42. Wang, L., and Pumera, M. (2021) Recent advances of 3D printing in analytical chemistry: Focus on microfluidic, separation, and extraction devices. TrAC Trends Anal. Chem., 135, 116151. 43. Agrawaal, H., and Thompson, J.E. (2021) Additive manufacturing (3D printing) for analytical chemistry. Talanta Open, 3, 100036.
44. Gross, B., Lockwood, S.Y., and Spence, D.M. (2017) Recent Advances in Analytical Chemistry by 3D Printing. Anal. Chem., 89 (1), 57–70.
45. Nesterenko, P.N. (2020) 3D printing in analytical chemistry: current state and future. Pure Appl. Chem., 92 (8), 1341–1355.
46. Mustapha, K.B., and Metwalli, K.M. (2021) A review of fused deposition modelling for 3D printing of smart polymeric materials and composites. Eur. Polym. J., 156, 110591.
47. Yadav, D., Chhabra, D., Kumar Garg, R., Ahlawat, A., and Phogat, A. (2020) Optimization of FDM 3D printing process parameters for multi-material using artificial neural network. Mater. Today Proc., 21, 1583–1591.
48. Tümer, E.H., and Erbil, H.Y. (2021) Extrusion-Based 3D Printing Applications of PLA Composites: A Review. Coatings, 11 (4), 390.
49. Azad, M.A., Olawuni, D., Kimbell, G., Badruddoza, A.Z.M., Hossain, Md.S., and Sultana, T. (2020) Polymers for Extrusion-Based 3D Printing of Pharmaceuticals: A Holistic Materials–Process Perspective. Pharmaceutics, 12 (2), 124.
50. Tan, D., Maniruzzaman, M., and Nokhodchi, A. (2018) Advanced Pharmaceutical Applications of Hot-Melt Extrusion Coupled with Fused Deposition Modelling (FDM) 3D Printing for Personalised Drug Delivery. Pharmaceutics, 10 (4), 203.
51. Melocchi, A., Uboldi, M., Cerea, M., Foppoli, A., Maroni, A., Moutaharrik, S., Palugan, L., Zema, L., and Gazzaniga, A. (2020) A Graphical Review on the Escalation of Fused Deposition Modeling (FDM) 3D Printing in the Pharmaceutical Field. J. Pharm. Sci., 109 (10), 2943–2957.
52. Chen, G., Xu, Y., Chi Lip Kwok, P., and Kang, L. (2020) Pharmaceutical Applications of 3D Printing. Addit. Manuf., 34, 101209.
53. Pires, F.Q., Alves-Silva, I., Pinho, L.A.G., Chaker, J.A., Sa-Barreto, L.L., Gelfuso, G.M., Gratieri, T., and CunhaFilho, M. (2020) Predictive models of FDM 3D printing using experimental design based on pharmaceutical requirements for tablet production. Int. J. Pharm., 588, 119728.
54. Sadia, M., Sosnicka, A., Arafat, B., Isreb, A., Ahmed, ´ W., Kelarakis, A., and Alhnan, M.A. (2016) Adaptation of pharmaceutical excipients to FDM 3D printing for the fabrication of patient-tailored immediate release tablets. Int. J. Pharm., 513 (1–2), 659–668.
55. Sadia, M., Arafat, B., Ahmed, W., Forbes, R.T., and Alhnan, M.A. (2018) Channelled tablets: An innovative approach to accelerating drug release from 3D printed tablets. J. Controlled Release, 269, 355–363.
56. Nasereddin, J.M., Wellner, N., Alhijjaj, M., Belton, P., and Qi, S. (2018) Development of a Simple Mechanical Screening Method for Predicting the Feedability of a Pharmaceutical FDM 3D Printing Filament. Pharm. Res., 35 (8), 151.
57. Mania, S., Ryl, J., Jinn, J.-R., Wang, Y.-J., Michałowska, A., and Tylingo, R. (2019) The Production Possibility of the Antimicrobial Filaments by Co-Extrusion of the PLA Pellet with Chitosan Powder for FDM 3D Printing Technology. Polymers, 11 (11), 1893.
58. Ranganathan, S.I., Kohama, C., Mercurio, T., Salvatore, A., Benmassaoud, M.M., and Kim, T.W.B. (2020) Effect of temperature and ultraviolet light on the bacterial kill effectiveness of antibiotic-infused 3D printed implants. Biomed. Microdevices, 22 (3), 59.
59. Thavornyutikarn, B., Lertwimol, T., Kosorn, W., Hankamolsiri, W., Nampichai, N., and Janvikul, W. (2021) 3DPRINTING antibacterial composite filaments containing synergistic antibacterial activity of green tea and tannic acid. Polym. Adv. Technol., 32 (12), 4733–4744.
60. Abudula, T., Qurban, R.O., Bolarinwa, S.O., Mirza, A.A., Pasovic, M., and Memic, A. (2020) 3D Printing of Metal/Metal Oxide Incorporated Thermoplastic Nanocomposites With Antimicrobial Properties. Front. Bioeng. Biotechnol., 8, 568186.
61. Bayraktar, I., Doganay, D., Coskun, S., Kaynak, C., Akca, G., and Unalan, H.E. (2019) 3D printed antibacterial silver nanowire/polylactide nanocomposites. Compos. Part B Eng., 172, 671–678.
62. Singh, D.D., Mahender, T., and Shashavali, S. (2021) Design, analysis and development of antimicrobial ventilator splitters for four patients. 050028.
63. Antony, S., Cherouat, A., and Montay, G. (2020) Fabrication and Characterization of Hemp Fibre Based 3D Printed Honeycomb Sandwich Structure by FDM Process. Appl. Compos. Mater., 27 (6), 935–953.
64. Mazzanti, V., Malagutti, L., and Mollica, F. (2019) FDM 3D Printing of Polymers Containing Natural Fillers: A Review of their Mechanical Properties. Polymers, 11 (7), 1094.
65. Fafenrot, S., Grimmelsmann, N., Wortmann, M., and Ehrmann, A. (2017) Three-Dimensional (3D) Printing of Polymer-Metal Hybrid Materials by Fused Deposition Modeling. Materials, 10 (10), 1199.
66. Kariz, M., Sernek, M., Obucina, M., and Kuzman, M.K. ´ (2018) Effect of wood content in FDM filament on properties of 3D printed parts. Mater. Today Commun., 14, 135–140.
67. Przekop, R.E., Kujawa, M., Pawlak, W., Dobrosielska, M., Sztorch, B., and Wieleba, W. (2020) Graphite Modified Polylactide (PLA) for 3D Printed (FDM/FFF) Sliding Elements. Polymers, 12 (6), 1250.
68. Zhao, H., Liu, X., Zhao, W., Wang, G., and Liu, B. (2019) An Overview of Research on FDM 3D Printing Process of Continuous Fiber Reinforced Composites. J. Phys. Conf. Ser., 1213 (5), 052037.
69. Gnanasekaran, K., Heijmans, T., van Bennekom, S., Woldhuis, H., Wijnia, S., de With, G., and Friedrich, H. (2017) 3D printing of CNT- and graphene-based conductive polymer nanocomposites by fused deposition modeling. Appl. Mater. Today, 9, 21–28.
70. Colella, R., Chietera, F.P., and Catarinucci, L. (2021) Analysis of FDM and DLP 3D-Printing Technologies to Prototype Electromagnetic Devices for RFID Applications. Sensors, 21 (3), 897.
71. Leigh, S.J., Bradley, R.J., Purssell, C.P., Billson, D.R., and Hutchins, D.A. (2012) A Simple, Low-Cost Conductive Composite Material for 3D Printing of Electronic Sensors. PLoS ONE, 7 (11), e49365.
72. Hamzah, H.H., Shafiee, S.A., Abdalla, A., and Patel, B.A. (2018) 3D printable conductive materials for the fabrication of electrochemical sensors: A mini review. Electrochem. Commun., 96, 27–31.
73. João, A.F., Rocha, R.G., Matias, T.A., Richter, E.M., Flávio S. Petruci, J., and Muñoz, R.A.A. (2021) 3D-printing in forensic electrochemistry: Atropine determination in beverages using an additively manufactured graphene-polylactic acid electrode. Microchem. J., 167, 106324.
75. Kwok, S.W., Goh, K.H.H., Tan, Z.D., Tan, S.T.M., Tjiu, W.W., Soh, J.Y., Ng, Z.J.G., Chan, Y.Z., Hui, H.K., and Goh,K.E.J. (2017) Electrically conductive filament for 3D-printed circuits and sensors. Appl. Mater. Today, 9, 167–175. 76. Zhang, D., Chi, B., Li, B., Gao, Z., Du, Y., Guo, J., and Wei, J. (2016) Fabrication of highly conductive graphene flexible circuits by 3D printing. Synth. Met., 217, 79–86.
77. Ambrosi, A., Shi, R.R.S., and Webster, R.D. (2020) 3Dprinting for electrolytic processes and electrochemical flow systems. J. Mater. Chem. A, 8 (42), 21902–21929.
78. Silva, A.L., Salvador, G.M. da S., Castro, S.V.F., Carvalho, N.M.F., and Munoz, R.A.A. (2021) A 3D Printer Guide for the Development and Application of Electrochemical Cells and Devices. Front. Chem., 9, 684256.
79. Pei, M., Shi, H., Yao, F., Liang, S., Xu, Z., Pei, X., Wang, S., and Hu, Y. (2021) 3D printing of advanced lithium batteries: a designing strategy of electrode/electrolyte architectures. J. Mater. Chem. A, 9 (45), 25237–25257. 80. Tijing, L.D., Dizon, J.R.C., Ibrahim, I., Nisay, A.R.N., Shon, H.K., and Advincula, R.C. (2020) 3D printing for membrane separation, desalination and water treatment. Appl. Mater. Today, 18, 100486.
81. Fijoł, N., Abdelhamid, H.N., Pillai, B., Hall, S.A., Thomas, N., and Mathew, A.P. (2021) 3D-printed monolithic biofilters based on a polylactic acid (PLA) – hydroxyapatite (HAp) composite for heavy metal removal from an aqueous medium. RSC Adv., 11 (51), 32408–32418.
82. Saidulu, D., Srivastava, A., and Gupta, A.K. (2022) Enhancement of wastewater treatment performance using 3D printed structures: A major focus on material composition, performance, challenges, and sustainable assessment. J. Environ. Manage., 306, 114461.
83. Zheng, Y., Sun, X., Liu, X., Xia, X., Xiao, L., Cao, C., Qian, Q., and Chen, Q. (2021) Improving the removal efficiency of methylene blue on 3D-printed camellia seed powder scaffold using porogen. Ind. Crops Prod., 171, 113930. 84. Kreider, M.C., Sefa, M., Fedchak, J.A., Scherschligt, J., Bible, M., Natarajan, B., Klimov, N.N., Miller, A.E., Ahmed, Z., and Hartings, M.R. (2018) Toward 3D printed hydrogen storage materials made with ABS-MOF composites. Polym. Adv. Technol., 29 (2), 867–873.
85. Saggiomo, V. (2020) 3D Printed Devices for Catalytic Systems, in Catalyst Immobilization, 1ed., Wiley, pp. 369–408.
86. Hurt, C., Brandt, M., Priya, S.S., Bhatelia, T., Patel, J., Selvakannan, Pr., and Bhargava, S. (2017) Combining additive manufacturing and catalysis: a review. Catal. Sci. Technol., 7 (16), 3421–3439.
87. Parra-Cabrera, C., Achille, C., Kuhn, S., and Ameloot, R. (2018) 3D printing in chemical engineering and catalytic technology: structured catalysts, mixers and reactors. Chem. Soc. Rev., 47 (1), 209–230.
88. Zhu, J., Wu, P., Chao, Y., Yu, J., Zhu, W., Liu, Z., and Xu, C. (2022) Recent advances in 3D printing for catalytic applications. Chem. Eng. J., 433, 134341.
89. Zhou, X., and Liu, C. (2017) Three-dimensional Printing for Catalytic Applications: Current Status and Perspectives. Adv. Funct. Mater., 27 (30), 1701134.
90. Laguna, O.H., Lietor, P.F., Godino, F.J.I., and CorpasIglesias, F.A. (2021) A review on additive manufacturing and materials for catalytic applications: Milestones, key concepts, advances and perspectives. Mater. Des., 208, 109927.
91. du Preez, A., Meijboom, R., and Smit, E. (2022) LowCost 3D-Printed Reactionware for the Determination of Fatty Acid Content in Edible Oils using a Base-Catalyzed Transesterification Method in Continuous Flow. Food Anal. Methods.
92. Alimi, O.A., Bingwa, N., and Meijboom, R. (2019) Homemade 3-D printed flow reactors for heterogeneous catalysis. Chem. Eng. Res. Des., 150, 116–129.
93. Ambrosi, A., and Pumera, M. (2018) Multimaterial 3D-Printed Water Electrolyzer with Earth-Abundant Electrodeposited Catalysts. ACS Sustain. Chem. Eng., 6 (12), 16968–16975.
94. Muñoz, J., Redondo, E., and Pumera, M. (2021) Versatile Design of Functional Organic–Inorganic 3D-Printed (Opto)Electronic Interfaces with Custom Catalytic Activity. Small, 17 (41), 2103189.
95. Hock, S., Rein, C., and Rose, M. (2022) 3D-Printed Acidic Monolithic Catalysts for Liquid-Phase Catalysis with Enhanced Mass Transfer Properties. ChemCatChem.
96. Díaz-Marta, A.S., Yañez, S., Lasorsa, E., Pacheco, P., Tubío, C.R., Rivas, J., Piñeiro, Y., Gómez, M.A.G., Amorín, M., Guitián, F., and Coelho, A. (2020) Integrating Reactors and Catalysts through Three-Dimensional Printing: Efficiency and Reusability of an Impregnated Palladium on Silica Monolith in Sonogashira and Suzuki Reactions. ChemCatChem, 12 (6), 1762–1771.
97. Sangiorgi, A., Gonzalez, Z., Ferrandez-Montero, A., Yus, J., Sanchez-Herencia, A.J., Galassi, C., Sanson, A., and Ferrari, B. (2019) 3D Printing of Photocatalytic Filters Using a Biopolymer to Immobilize TiO 2 Nanoparticles. J. Electrochem. Soc., 166 (5), H3239–H3248.
98. Wang, Y., Gawedzinski, J., Pawlowski, M.E., and Tkaczyk, T.S. (2018) 3D printed fiber optic faceplates by custom controlled fused deposition modeling. Opt. Express, 26 (12), 15362.
99. Kool, L., Bunschoten, A., Velders, A.H., and Saggiomo, V. (2019) Gold nanoparticles embedded in a polymer as a 3D-printable dichroic nanocomposite material. Beilstein J. Nanotechnol., 10, 442–447.
100. Kool, L., Dekker, F., Bunschoten, A., Smales, G.J., Pauw, B.R., Velders, A.H., and Saggiomo, V. (2020) Gold and silver dichroic nanocomposite in the quest for 3D printing the Lycurgus cup. Beilstein J. Nanotechnol., 11, 16–23. 101. Gleadall, A. (2021) FullControl GCode Designer: Open-source software for unconstrained design in additive manufacturing. Addit. Manuf., 46, 102109. 102. Baas, S., and Saggiomo, V. (2021) Ender3 3D printer kit transformed into open, programmable syringe pump set. HardwareX, 10, e00219.
103. Ponzetti, M., Devarapu, G.C.R., Rucci, N., Carlone, A., and Saggiomo, V. (2022) HistoEnder: a 3D printer-based histological slide autostainer that retains 3D printer functions.
104. Koch, F., Thaden, O., Tröndle, K., Zengerle, R., Zimmermann, S., and Koltay, P. (2021) Open-source hybrid 3D-bioprinter for simultaneous printing of thermoplastics and hydrogels. HardwareX, 10, e00230.
AWK是一個古老的Unix程式,在1970年代在當時資通訊最前沿的貝爾實驗室,由三位工程師Alfred Aho, Peter Weinberger, Brian Kernighan共同開發的,主要是一款字串處理程式。
AWK reads the input a line at a time. A line is scanned for each pattern in the program, and for each pattern that matches, the associated action is executed.