原始論文:
Cole Trapnell,Adam Roberts,Loyal Goff,Geo Pertea,Daehwan Kim,David R Kelley,Harold Pimentel,Steven L Salzberg,John L Rinn& Lior Pachter. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks.Nature Protocols.7,562–578(2012)Doi:10.1038/nprot.2012.016
RNAseq的資料分析主要有兩個大目的:
- 找尋新的基因或是transcripts
- 比較不同狀態下的基因表現亮
而在分析步驟上,可以把它拆成三個分析流程:
- read alignment
- transcript assembly 或是 genome annotation
- transcript and gene quantification
本篇論文主要介紹用期開發的程式作為workflow,並且簡單介紹每個tool之間的異同。
所使用的工具和工作流程:
Read alignment with Tophat
Reads拼回去reference的步驟非常重要,他可以找出所有可能的insertion/deletion/indel,這些資訊可以讓我們了解辨識出相對於對照序列的polymorphism,另外,對不回去的reads,可能是新的protein-coding genes或是noncoding RNA。這邊拼回去的結果,也會影響之後如何去計算transcript abundance的數值。

Transcripts assembly with Cufflinks
為了要知道各種基因的表現量,我們必須知道各種reads所對應的isoforms transcripts,這邊就必須把Reads拼組成各種特定的transcipts,這步驟牽涉到如何辨別不同種transcripts variant ,所以最後產生transfrags來表達所有可能。
cufflinks會將一些low abundance的tansfrag當作是來自於immature transcripts而丟棄,這部分是要了解的。(或許這步驟可以思考一下!),有時候我們會將所有的bam pool在一起讓他找transcripts,但這種作法其實一方面增加電腦計算的loading,另一方面,則是會讓程式在計算isoform時增加很多變數,所以cuffmerge應映兒生,他可以把individual產出的transfrag merge在一起看!
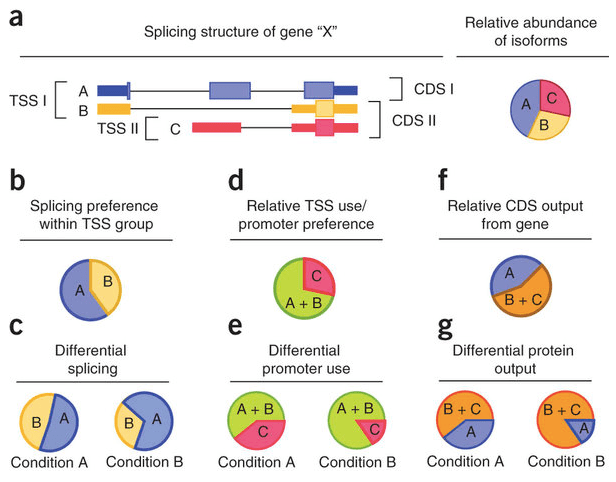
Differeial Expression Analysis with Cuffdiff
cuffdiff可以輸出好幾個檔案描述不同基因和transcripts的表現量差別和p value以及其名字和在基因體上的位置,另外,cuffdiff也可以把同一組基因裡面不同TSS的transcripts分組去計算他們之間比例的變化!
視覺化資料CummeRbound
cuffdiff提供基因和transcripts的表現量分析,且這些資料是以tab-delimited的方式輸出很方便後續的分析,而CummeRbound則是提供一個更容易操作的視覺化分析,可以將cuffdiff的計算結果做更細緻的分析。
替代工具:
- Read-alignment
- GSNAP
- START
- Map-Splice
- Transcriptome reconstruction
- De novo transcripts assembly
- Trinity
- Trans-Abyss
- Oases
- De novo transcripts assembly
- Quantification
- Differential expression
有趣的相關論文:
Read-alignment
- & Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
- et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010).
- , , , & Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 38, 4570–4578 (2010).
Transcript reconstruction
- et al. Alternative expression analysis by RNA sequencing. Nat. Methods 7,843–847 (2010).
Quantification
- , , , & RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500 (2010).
- , , & Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015 (2010).
- , , & Estimation of alternative splicing isoform frequencies from RNA-seq data. Algorithms Mol. Biol. 6, 9 (2011).
Differential Expression
- & Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
- , & edgeR: a Bioconductor package for differential expression analysis of digital gene expression data.
- Bioinformatics 26, 139–140(2009)., , , & DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).