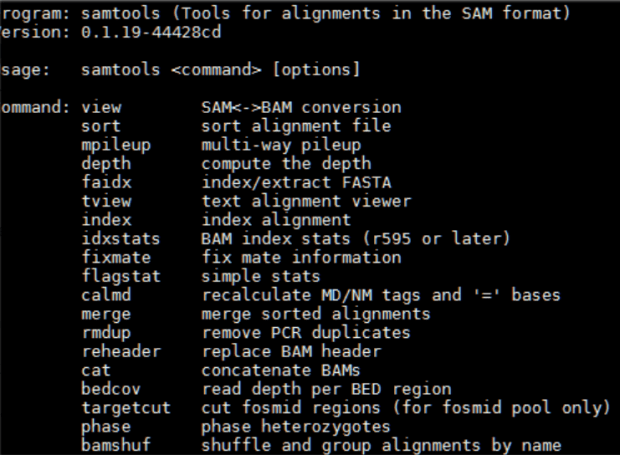
samtools是在inspect或是處理bam/sam黨非常重要的工具,這邊簡單介紹他的功能和基本語法使用。
基本上,samtools對於sam/bam檔案有三大功能:
第一 ,就是檔案格式的轉換(bam to sam/sam to bam)
第二,sam/bam reads的篩選,過濾、查看
第三,進階的variant calling分析(mpileup)
 一 、檔案格式的轉換
一 、檔案格式的轉換
我們可以使用samtools將bam檔儲存成sam黨,反之亦然,雖然tools一開始的defaut是input bam,但都是可以調整的。
bam 轉 sam: $samtools view input.bam >output.sam
sam 轉 bam: $samtools view -b input.sam > output.bam
二、篩選,過濾、查看sam/bam
排序bam檔案: $samtools sort -f input.bam output.bam
索引bam檔案: $samtools index input.bam (會跑出 *.bai 檔案 )
通常再匯入IGV或是某些軟體前會需要進行這兩個步驟
過濾特定flag的reads
留下特定flag的reads $samtools view -f [FLAG value]
濾掉特定flag的reads $samtools view -F [FLAG value]
(關於samformat的flag可參考此)
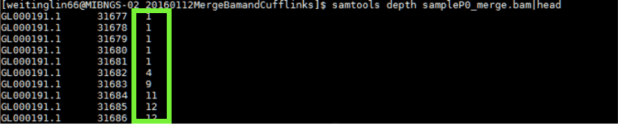
可以看alignment的depth和reads mapping status,這功能蠻好用的!
看每個reference序列定序深度
$ samtools depth data.bam
綠色框框代表每個序列對應到的reads疊加數
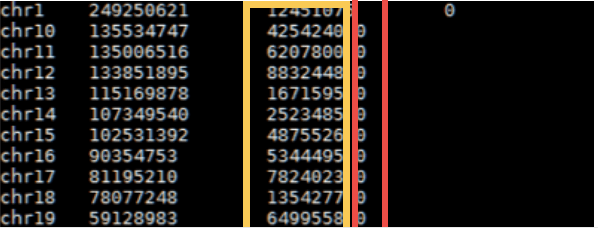
$samtools idxstates data.bam
黃色代表是左邊染色體上有被mapped上的reads數量,右邊代表的是unmapped量
第三種功能則是calling variant

這邊就需要調整比較多參數,其實這些功能蠻方便的,可以快速地瞭解整體bam檔的狀態!

在〈samtools 使用〉中有 1 則留言