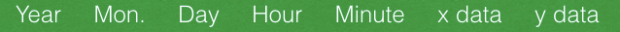RNAseq資料處理的步驟非常繁複,而第一步就是要看定序出來的品質如何,品質不好的話是否要去掉差的定序reads,特意設計疊加的reads是否要先處理完後再繼續下一步的aligment、是否有被污染、過多的adaptors(比較短的library中容易碰到的問題)。
最常見用來評估定序品質的工具為FastQC
FASTQC 是一款用來分析RNA-seq FASTQ 檔案的軟體,可以在command line環境下執行.
分析後會取得的資料:
- 檔案名稱:最原始分析前之檔案名稱
- 檔案類型
- 解碼(encoding):其品質數值所表示的ASCII
- 全部定序數量(Total Sequence):總共有多少定序片段
- 過濾定序(filtered sequence):在Casava模式下,在分析過程中片段序列被標誌且移除
- 定序長度(Sequence Length):最短和最長的定序序列
- GC比例(GC content)
FastQC 執行完,會給出11張圖:
Per Base Sequence Quality 每個base平均品質

- 分析內容
- 可以用來看是否有特別低品質的定序序列,通常都是因為照向品質不好所造成的,像是位在定序晶片邊緣的點
- 達不同品質指標,圖標會顯示之圖例
- 警告(Warming):當平均品質小於27/0.2% error rate
- 失敗(Failure):當平均品質小於20/1% error rate
- 備註:經過quality trimming後可能會有偏差,或是看不出error batch?
Per Sequence Quality Scores 每條序列中不同位點的品質
- 分析內容
- 顯示序列裡不同位置平均品質範圍(只要是fastq中之資料)
- BoxWhisker type Plot圖中標示的解釋:
- 中間紅色線:中間值(median)
- 黃色長條:四分位距內的值(25-75%)
- 上下whiskers: 各代表10%和90%的點
- 藍色線:代表平均值(mean)
- 背景色
- 綠色very good quality
- 橘色reasonable quality
- 紅色poor quality
- 標題上是FastQC猜測此定序所使用的方式,在虛列品質好時比較容易猜測錯誤.
- 達不同品質指標,圖標會顯示之圖例
- 警告(Warming):當25%之任何序列位點<10或是median值<25
- 失敗(Failure):當25%之任何序列位點<5或是median值<20
- 問題導因:
- 原因一: 跑定序的時間過長,造成後面定序出來的品質下降
- 處理方式: Quality Trimming(依據平均品質來決定裁掉的長度)
- 通常在adapter read-through也會有問題
- 原因二:在一開始的時候就產生問題,可能有泡泡跑過定序的夾層,這時候就不適合trim,會因此拋棄過多好品質的序列
- 原因三:太低的coverage可能會造成false negative
- 原因一: 跑定序的時間過長,造成後面定序出來的品質下降
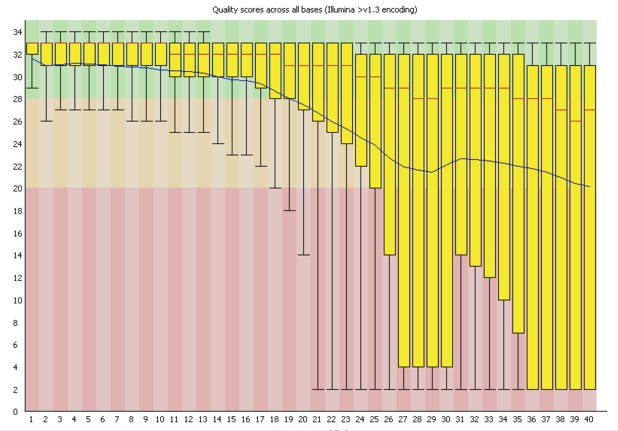
Per Base Sequence Content序列中不同位置其ATCG所佔的比例
- 分析內容:
- 在一個隨機的library中,其序列中不同base應該不會有太大的差別.
- 通常在開頭的地方容易有不平均的ATCG比例,通常在Library製備過程中priming使用到random hexamers(幾乎所有RNA-seq的製備都會使用),或是在fragmentation過程中使用transposases,通常會在reads開頭有偏差出現,使得在有enrichment在5’端的different k-mers處,這種技術性造成的偏差無法藉由trimming消掉,但在downstream analysis中也不會造成太大的問題
- 達不同品質指標,圖標會顯示之圖例
- 警告(Warming):當任何一處的ATCG其差異超過10%
- 失敗(Failure):當任何一處的ATCG其差異超過20%
- 問題導因:
- 原因一:Overrepresented sequence
- 可能是做定序所使用的adapter dimers或是rRNA
- 原因二:Biases Fragmentation
- 因製備Library中所使用的random hexamers在前12bases會有selected biase,但這在後續的process可以處理掉,不太會影響後續的分析
- 原因三:Biases Composition Libraries
- 大多數的library本身就會有些composition的偏差,尤其是使用sodium bisulphite製備的,其會造成cytosine變成thymines,所以會使得C的比值偏高
- 原因四:Extreme Trimming
- 在尾端會出現,因為很積極地將跟adapter match的序列去除掉
- 原因一:Overrepresented sequence
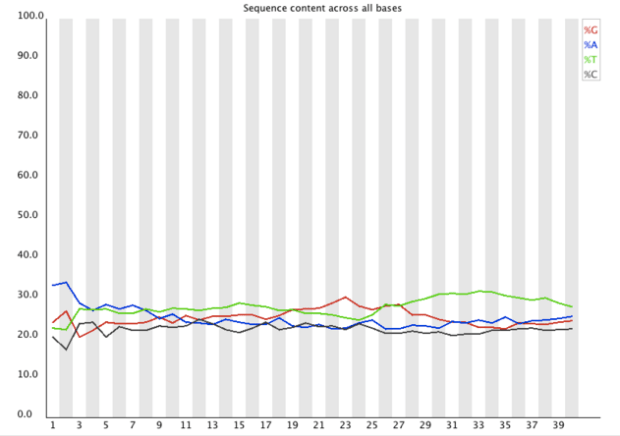
Per Sequence GC Content 測量GC在序列中的比例
- 分析內容:
- 其主要是分析不同序列的GC之分布圖,應該要跟理論分布差不多
- 比較陡或是尖:可能是被adapter汙染
- 比較寬:可能是被不同物種所污染
- 達不同品質指標,圖標會顯示之圖例:
- 警告:偏離理論值15%
- 失敗:偏離理論值30%
Per Base N Content 平均每個位點壞品質(N)

- 分析內容:
- 當某個序列即位點定序品質很差,則會用N代表
- 通常當某個部分其N所佔的比例上升的話,其會使後面的分析無法獲得有效的base cal.
- 達不同品質指標,圖標會顯示之圖例:
- 警告:任一個位置其N所佔比例大於5%
- 失敗:任一個位置其N所佔比例大於20%
- 備註原因:
- 原因一:
- 通常是整體定序品質不佳,可以檢查一下特定bin的coverage,可能有某一個bin其定到的序列非常少
- 原因二:
- 有嚴重偏差的LIBRARY,在Per Base Sequence Content也可以看出來
- 原因一:
Sequence Length Distribution 定序長度分布圖

- 分析內容:
- 顯示平均定序的長度,這部分就要先了解是使用何種定序方式,有的會有單一長度,有的則是不同
- 達不同品質指標,圖標會顯示之圖例:
- 警告:假如定序的長度有不一樣的話
- 失敗:任一個定序長度是零
Duplicate Sequences 重複序列

- 分析內容:
- 在一個均勻的library中序列應該只會出現一次,很低程度的重複率可能暗示高coverage,高度序列重複則可能是library中有某種程度的enrichment bias
- 為了使分析的計算效率提高,這個分析模組只會分析在前100000的序列,另外在超過10個重複的序列,其會被放在一起呈現
- 比較長的定序讀數,通常會造成一定程度的低估重複率
- 藍色線:代表完整序列其重複率, 紅色線:代表去掉重複後不同序列的比例
- Overrepresented Sequences
- Adapter Content
- Kmer Content
- Per Tile Sequence Quality
去掉品質差Reads的工具:Trimmomatic
可以用來處理illumina機器裡的adaptor,其也可以將low-quality的base或是N base去掉,其可以處理single-end和paired-end的資料。此程式是java base的軟體,在command line下操作。
處理特殊設計的paired-end 實驗設計:BBMerge 和 FLASH
有些實驗設計時,會將paired-end read設計成疊在一起的樣子,而這兩款程式可以將重疊的paired-end reads合成單一條較長的reads,且能計算出innert-size的分布圖。
偷偷查了一下FLASH的使用趨勢,似乎暴增人數使用!