SRA database (Sequence Read Archive)是目前次世代定序實驗資料原始檔案的儲存地方,在越來越追求研究reproducible的時代,大多數基因體研究都會要求將資料上傳到雲端,讓其他科學家也能使用, 所以看到跟自己相關且興趣的基因體相關論文,想要自己下載其實驗定序出來的資料來分析一下,便可以先來NIH的網站瀏覽一番,看有無自已有興趣的資料,在用其原始資料accession number來下載,但次世代定序的資料動輒好幾GB,明智的做法是使用公用的linux server去下載,此使可以利用NIH 開發的SRA toolkit,這軟體主要是在command line環境下操作,只要知道自己想要下載資料的accession number,便能用一行指令下載好幾gb的檔案,甚至可以進行簡單的檔案轉換處理,而只要使用SRA toolkit便能簡單做到,這邊介紹一下如何用SRA toolkit來下載心儀的資料。
在此之前,我們先來了解一下SRA database其對檔案命名的眉角,其實是有一定的邏輯的,大致如下的命名編號架構(這邊指的是accession number):
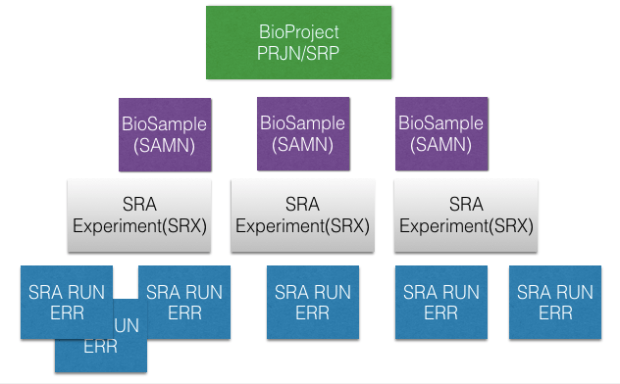
簡單來說,每個研究都會有個計畫編號,開頭大多會是PRJN或是SRP,這種計畫通常都會不只做一組檢體,而每個送去做定序的檢體都會有專屬編號,以SAMN開頭,而此檢體使用的定序實驗設計,也會有個編號,以SRX開頭,裡面會記錄此檢體所使用的定序機器種類、library type、定序設計(paired-end, single ….),而在同一次定序中其跑的run也會有編號,以SRR開頭。
接下來寫一下,SRA toolkit的下載和使用:
第一步:到此頁面下載符合自己系統的版本 link

第二步:在系統下解壓縮
這邊以linux/mac系統的指令為例
$tar -xzf sratoolkit.current-centos_linux64.tar.gz
第三步:設定至系統路徑
此tool解壓縮完,其實就可以使用了,其tools的執行擋放在/sratoolkit/bin/的目錄下,直接打想要的tool linux是無法直接啟動,所以要去修改bash_profile,參考這篇網誌 如何設定檔案室系統路徑變數中
第四步:常用tools
其中最常用的工具就是fastq-dump了,可以用其直接使用accession number來query NIH 的SRA database並且下載fastq檔案,這邊是其指令介紹:
主要指令依功能分成三大類:
- data formating:下載後的檔案希望為什麼樣的格式,可以為fasta、fastq、sam,可以存成一個檔案,或是每個reads分開存
- filtering:指定要下載特定區間內的reads
- workflow and piping:檔案最後要直接顯示在螢幕、或是儲存到特定的目錄中