在之前有討論過RNAseq的分析中,基因表現的分析有很多不同的策略,一種採用定序片段(reads)對應到基因區域的數量代表基因表現(counting reads per genes),另一種則是使用RPKM(Reads Per Kilobase per Million mapped reads) ,兩種方式都有相對應針對資料正規化(normalization)的想法,可以參考這篇。
HTSeq 是用來處理alignment好的bam檔,將其產生counting reads table。而之後便可以把這資料丟到DESeq做基因表現分析(gene expression analysis),DESeq是R語言的package,可以用來做基因表現分析,其中用的原理便是採用counting reads per genes來做分析。
首先,HTSeq是一款python的軟體,除了計算reads count per genes 也可以用來處理基本的bam file writing 或是genomic interval的處理,其必須搭配安裝python 的一些週邊package,如PyPI、pysam,在MacOS X 上則先安裝xcode、MacPort(套件管理程式,用來安裝SciPy的套件),另外要注意其支援的為python2.7版本。
在使用htseq-count時最重要的是他用三種mode,關於他如何定義reads對到genes的關係,一般defaut為union。官網說明連結
使用counting reads功能的簡單語法如下:

基本上,輸入的bam檔必須經過sorting,最好是by reads names的sort setting,可以節省記憶體的要求,另外,annotation上,使用ensembl的annotation其gene ID和transcripts ID有區別性,對於程式來說比較好。
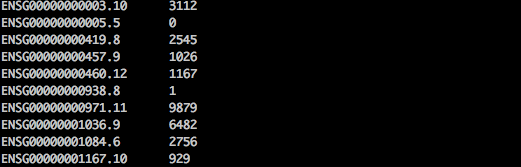
輸出會呈現如下: