Han Y, Gao S, Muegge K, Zhang W, Zhou B. (2015) Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights 9(Suppl 1):29-46. [article]
計算好基因的表達量,之後的data normalization其實是很重要的一環,這一步驟要考慮的細節很多,像是transcripts size、GC content、sequencing depth、sequencing error rate、insert size。
- Li S, Łabaj PP, Zumbo P, et al. Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat Biotechnol. 2014;32(9):888–95.
- Filloux C, Cédric M, Romain P, et al. An integrative method to normalize RNA-seq data. BMC Bioinformatics. 2014;15:188.
在評估各種標準化的方式,可以使用measurement error model來評估其標準化的效力。
- Ager-Wick E, Henkel CV, Haug TM, Weltzien FA. Using normalization to resolve RNA-Seq biases caused by amplification from minimal input. Physiol Genomics. 2014;46(21):808–20
- Sun Z, Zhu Y. Systematic comparison of RNA-Seq normalization methods using measurement error models. Bioinformatics. 2012;28(20):2584–91.
目前有很多標準化的方式,像是quantile normalization,這方法在處理microarray資料上很有效,在RNAseq中能提高data quality,即使是low amount的RNA。
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics.
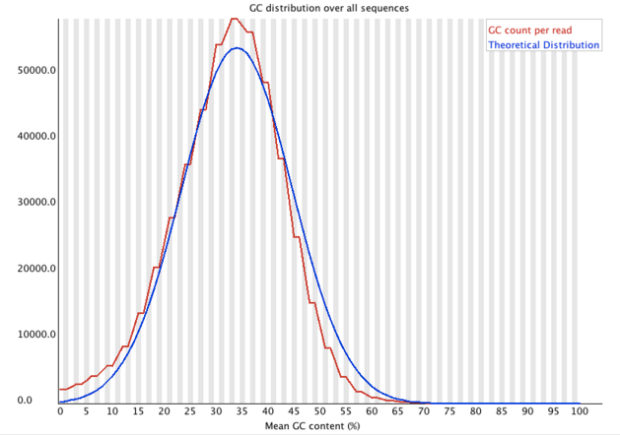
在R語言中的EDASeq,其先使用within-lane normalization再來between-lane normalization,可以有效降低GC-content所造成的問題
- Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-seq data. BMC Bioinformatics. 2011;12:480.
Lowess normalization的方法在microRNA 的資料處理蠻有效的。
- Garmire LX, Subramaniam S. Evaluation of normalization methods in mammalian microRNA-Seq data. RNA. 2012;18(6):1279–88.
目前在處理RNAseq資料上的normalization依舊需要有更好的方法被發展出來。