常見的Alignment Genomic Data Archive Format
alignment後的基因定序資料通常是任何分析的起頭,如廚師把配菜弄好擺在流理台後,要大顯身手,此時一個好的工具來處理就是非常重要,不然會有種看者滿山資料卻不知從何下手。alignment後的定序資料通常就是已有配對到reference genome上的reads,目前此類檔案有三種format: SAM, BAM, CRAM,檔案大小依序也是從左到右,而SAM(Sequence Alignment/Map)和BAM(Binary Alignment/Map)兩個檔案基本上是同樣的內容,差別在SAM是人看得懂的,BAM是壓縮成binary的,前兩種檔案格式出現的時候,定序資料量還沒現在這麼大,最近連BAM格式還是被嫌太大,CRAM format是會逐漸取代前者的壓縮方式,EMBL已經改用CRAM的格式來儲存定序檔案了,其概念是所謂的reference 壓縮的模式,如下圖所事是: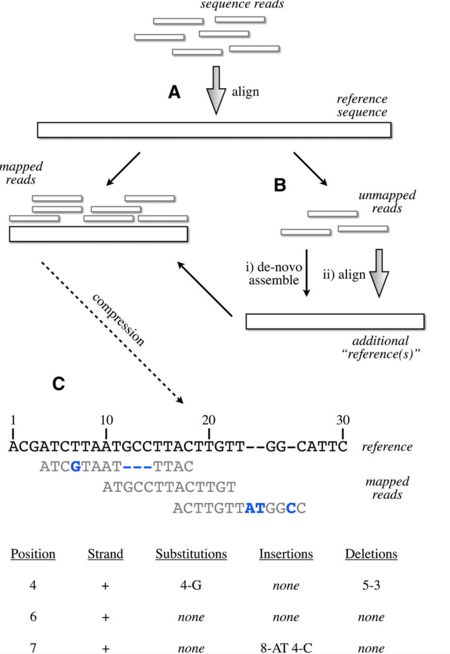
簡單來說,就是以reference上相對位置和改變的資訊來儲存,而非直接儲存reads序列,
整個降低檔案儲存需要的大小。使用cram格式相對於bam可以節省約10-30%的檔案大小,但因為cram的格式是基於reference來減低檔案格式,需要固定的reference版本,才不會再還原時候有問題,所以每個CRAM檔案會有MD5 checksum,來確保記錄所使用的reference版本。
關於CRAM的相關reference:
Hsi-Yang Fritz, et al. (2011). Genome Res. 21:734-740
Cochrane G. et al. (2012). GigaScience 1:2
SAM Format的形式
SAM的檔案架構分兩部分,一部分是Header,另一部分是Alignment。外觀如下圖:

Header的部分位在整個檔案的一開頭,裡面會有這份檔案的基本資料,如有無sorted過、裡頭使用的reference及其長度、使用過什麼工具處理和alignment時所下的指令。Header裡頭會有幾個關鍵組成:@SQ/ @RG/ @PG,分別代表以下的意義
@SQ 這個開頭含有做alignment所使用的reference序列資訊,SN代表的是sequence name,而緊跟者為LN,代表此參考序列的長度
@RG代表者read group的資訊和sample的基本資料,部分軟體會根據裡頭的ID,去辨識有無batch effect,有的會有PL指標去代表其所使用的定序平台
@PG裡頭含有此次alignment所使用的程式資訊,CL指標後面有所下的指令,VL後面有使用的軟體版本資訊
Alignment的部分就是實際一個個read的alignment資料,一行為一個reads,其用以下的資訊來說明每個reads的狀況:

依序是每個Reads獨一無二的編號,通常還有在定序機器上的位置,FLAG,則是代表此reads的mapping狀態,chr則很明顯的是對到的染色體,Start代表reads對應到reference上的開始位置,CIGAR則代表這個reads每一個位置跟reference mapping的狀態,是否100Match到,後面還有一些進階資訊,假如是paired-read則會顯示他的mate reads的位置。
因為sam檔沒有壓縮過,所以大多數會以bam檔形式儲存,必須使用samtools來轉換,以便閱讀上面那些資訊,這邊samtools在處理bam/sam檔案上就非常重要了!

在〈SAM, BAM and CRAM〉中有 1 則留言